

394 |
Growth and development |
|
|
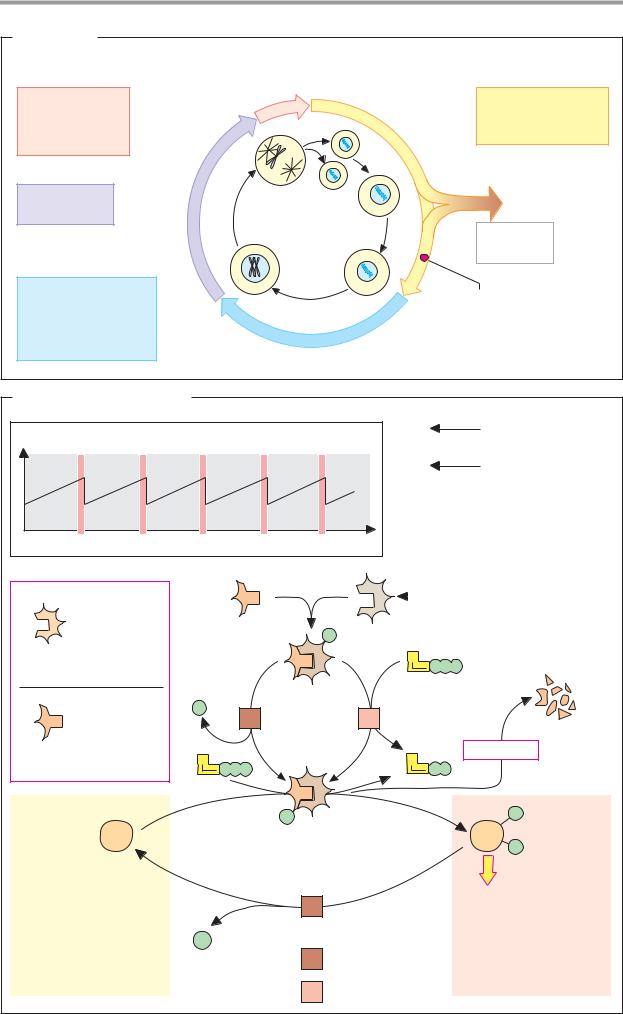
Cell cycle |
The above outline of cell cycle progression |
||
|
|
can be examined here in more detail using the |
|
A. Cell cycle |
G2–M transition as an example. |
||
Entry of animal cells into mitosis is based |
|||
|
|
||
Proliferating cells undergo a cycle of division |
on the “mitosis-promoting factor” (MPF). MPF |
||
(the cell cycle), which lasts approximately |
consists of CDK1 (cdc2) and cyclin B. The in- |
||
24 hours in mammalian cells in cell culture. |
tracellular concentration of cyclin B increases |
||
The cycle is divided into four different phases |
constantly until mitosis starts, and then de- |
||
(G1, S, G2, and M—in that sequence). |
clines again rapidly (top left). MPF is initially |
||
Fully differentiated animal cells only divide |
inactive, because CDK1 is phosphorylated and |
||
rarely. These cells are in the so-called G0 |
cyclin B is dephosphorylated (top center). The |
||
phase, inwhich they can remain permanently. |
M phase is triggered when a protein phos- |
||
Some G0 cells return to the G1 phase again |
phatase [1] dephosphorylates the CDK while |
||
under the influence of mitogenic signals |
cyclin B is phosphorylated by a kinase [2]. In |
||
(growth factors, cytokines, tumor viruses, |
its active form, MPF phosphorylates various |
||
etc.), and after crossing a control point (G1 to |
proteins that have functions in mitosis—e.g., |
||
S), enter a new cycle. DNA is replicated (see |
histone H1 (see p. 238), components of the |
||
p. 240) during the S phase, and new chroma- |
cytoskeleton such as the laminins in the nu- |
||
tin is formed. Particularly remarkable in mor- |
clear membrane, transcription factors, mitotic |
||
phological terms is the actual mitosis (M |
spindle proteins, and various enzymes. |
||
phase), in which the chromosomes separate |
When mitosis has been completed, cyclin B |
||
and two daughter cells are formed. The M and |
is marked with ubiquitin and broken down |
||
S phases are separated by two segments |
proteolytically by proteasomes (see p. 176). |
||
known as the G1 and G2 phases (the G stands |
Protein phosphatases then regain control |
||
for “gap”). In the G1 phase, the duration of |
and dephosphorylate the proteins involved |
||
which can vary, the cell grows by de novo |
in mitosis. This returns the cell to the inter- |
||
synthesis of cell components. Together, the |
phase. |
||
G1, G0, S, and G2 phases are referred to as |
|
||
the interphase, which alternates in the cell |
Further information |
||
cycle with the short M phase. |
|
||
|
|
The G1–S transition (not shown) is particu- |
|
B. Control of the cell cycle |
larly important for initiating the cell cycle. It |
||
is triggered by the CDK4–cyclin D complex, |
|||
|
|
||
The progression of the cell cycle is regulated |
which by phosphorylating the protein pRb |
||
by interconversion processes. In each phase, |
releases the transcription factor E2F previ- |
||
special Ser/Thr-specific protein kinases are |
ously bound to pRb. This activates the tran- |
||
formed, which are known as cyclin-depen- |
scription of genes needed for DNA replication. |
||
dent kinases (CDKs). This term is used be- |
If the DNA is damaged by mutagens or ion- |
||
cause they have to bind an activator protein |
izing radiation, the protein p53 initially delays |
||
(cyclin) in order to become active. At each |
entry into the S phase. If the DNA repair sys- |
||
control point in the cycle, specific CDKs asso- |
tem (see p. 256) does not succeed in remov- |
||
ciate with equally phase-specific cyclins. If |
ing the DNA damage, p53 forces the cell into |
||
there are no problems (e.g., DNA damage), |
apoptosis (see p. 396). The genes coding for |
||
the CDK–cyclin complex is activated by phos- |
pRb and p53 belong to the tumor-suppressor |
||
phorylation and/or dephosphorylation. The |
genes (see p. 398). In many tumors (see |
||
activated complex in turn phosphorylates |
p. 400), these genes are in fact damaged by |
||
transcription factors, which finally lead to |
mutation. |
||
the |
formation of the proteins that are re- |
|
|
quired in the cell cycle phase concerned (enzymes, cytoskeleton components, other CDKs, and cyclins). The activity of the CDK–cyclin complex is then terminated again by proteolytic cyclin degradation.
|
|
Cell proliferation |
395 |
A. Cell cycle |
|
|
|
M phase |
|
G1 phase |
|
Mitosis |
0 h |
RNA and protein |
|
|
|
||
Chromosome |
|
synthesis |
|
separation |
|
Cell growth |
|
Cell division |
|
|
|
G2 phase |
|
G0 phase |
|
Preparation |
|
|
|
for mitosis |
|
4 h |
|
12h |
|
|
|
|
|
No cell |
|
S phase |
|
division |
|
|
|
|
|
DNA replication |
|
Restriction |
|
Histone synthesis |
|
|
|
|
point |
|
|
Centrosome formed |
|
|
|
|
|
|
|
Chromosome |
8 h |
|
|
duplication |
|
|
|
|
|
|
B. Control of the cell cycle |
|
|
|
|
|
|
|
|
||
Cyclin B concentration |
|
|
|
M = Mitosis |
|
|
|
Reactions early |
||
|
|
|
|
|
|
in mitosis |
||||
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
Reactions late |
|
|
|
|
|
|
|
|
|
|
in mitosis |
|
M |
M |
M |
|
|
M |
Time |
|
|
|
|
|
|
|
|
|
|
|
Cyclin-dependent |
|
||
|
|
Cyclin B |
|
|
|
|
protein kinase (CDK1) |
|||
Cyclin- |
|
|
|
|
|
|
|
|
|
|
dependent |
Regulatory |
|
|
|
Catalytic |
|
|
|||
protein |
|
|
|
P |
subunit |
|
|
|||
|
subunit |
|
|
|
|
|
||||
kinases |
|
|
|
|
|
|
|
|
Cyclin |
|
|
|
|
|
|
|
A |
|
|
||
|
|
|
|
|
|
|
|
|
||
CDK 1 – 6 |
|
|
|
|
|
|
|
|
fragments |
|
|
|
|
|
|
|
P |
P |
P |
||
|
|
|
|
|
Inactive |
|
|
|
|
|
|
|
P |
|
|
protein |
|
|
|
|
|
Cyclin |
|
|
|
1 |
kinase |
2 |
|
|
|
|
Cyclins A – E |
|
A |
|
|
|
|
A |
|
Proteolysis |
|
|
|
|
|
|
|
|
|
|||
|
P |
P |
P |
|
|
P |
P |
|
|
|
|
|
|
|
|
|
|||||
Interphase |
|
|
|
|
P |
|
|
|
P |
Mitosis |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
MPF |
|
|
|
P |
|
Protein |
|
|
|
Active protein kinase |
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
Histone H1 |
|
|
|
|
1 |
|
|
|
Spindle formation |
|
Laminin |
|
|
|
|
|
|
|
Chromosome |
||
Protein kinases |
|
|
|
|
|
|
|
|
condensation |
|
Transcription |
|
P |
|
|
|
|
|
|
Disappearance of |
|
factors |
|
|
|
|
1 |
Phosphoprotein |
|
nuclear membrane |
||
|
|
|
|
|
|
phosphatase |
|
Transcription stop |
||
Other proteins |
|
|
|
|
2 |
Protein kinase |
|
Cyclin degradation |
||
|
|
|
|
|
|
|
|
|||

|
|
|
Cell proliferation |
397 |
|
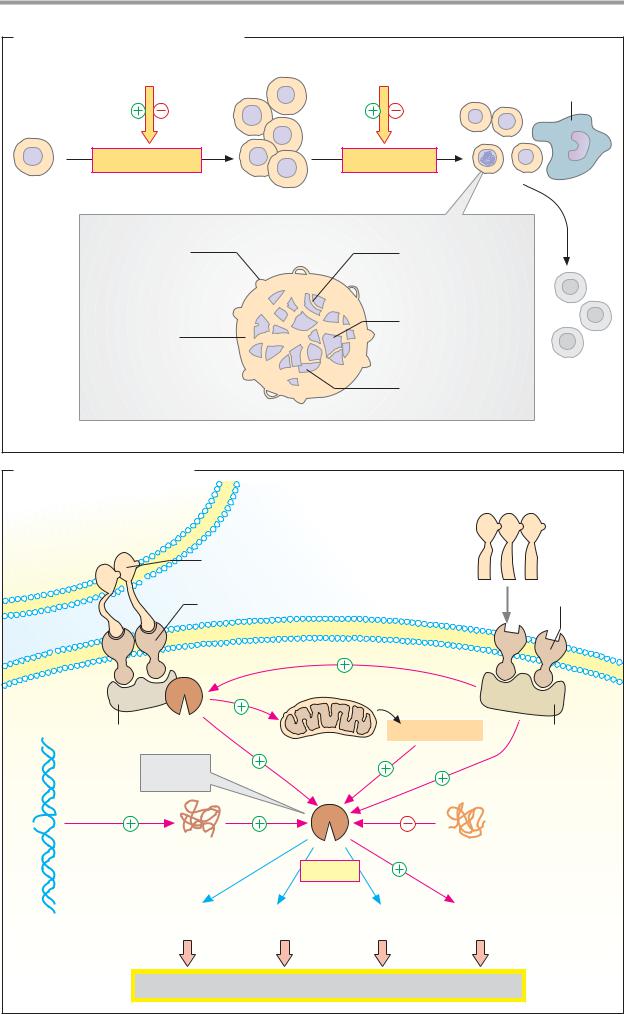
A. Cell proliferation and apoptosis |
|
|
|
|
|
Factors |
|
Factors |
|
Phagocytic |
|
|
|
|
|
macrophage |
|
Cell proliferation |
|
Apoptosis |
|
|
|
Constant cell number |
|
|
|
|
|
Changes in |
|
Dissolution |
|
|
|
membranes |
|
of nuclear structure |
|
||
Shrinking of |
|
Condensation |
|
|
|
cytoplasm |
|
of chromatin |
|
|
|
|
|
Fragmentation |
|
|
|
Apoptotic cell |
|
of DNA |
|
|
|
|
|
|
|
|
|
B. Regulation of apoptosis |
|
|
|
|
|
Cytotoxic T cell |
|
|
|
|
|
Fas ligand |
|
TNF-α |
TNF |
|
|
|
|
|
|
||
Fas |
|
|
|
receptor |
|
|
|
|
type I |
|
|
receptor |
|
|
|
|
|
|
|
|
|
|
|
Caspase 8 |
|
|
|
|
|
|
Mitochondrion |
|
|
|
|
FADD |
|
Cytochrome c |
TRADD |
||
Effector |
|
|
|
|
|
caspases |
|
|
|
|
|
p53 protein |
|
|
bcl-2 protein |
|
|
|
cleave |
|
|
|
|
Other proteins |
snRNA |
Laminin |
Caspase-activated |
|
|
DNA with |
proteins |
|
DNAase |
|
|
|
|
|
|
|
|
irradiation |
|
|
|
|
|
damage |
Apoptosis |
|
|
|
|
|
|
|
|
||


400 |
Growth and development |
|
|
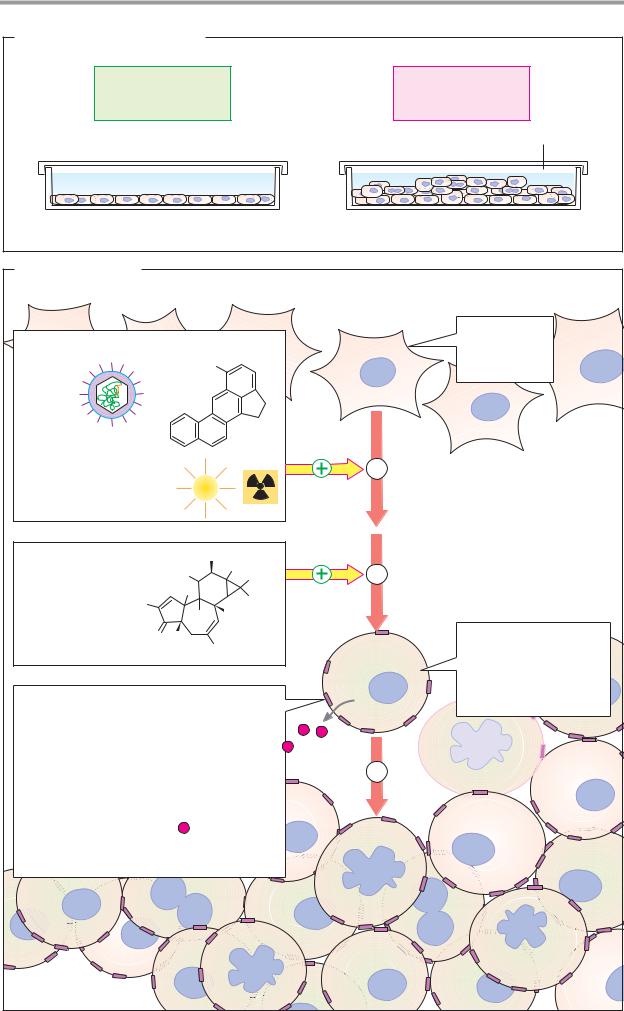
Tumors |
|
tumor cells (group 1) or are induced by them |
|
|
|
in other cells (group 2). Group 1 tumor |
|
A. Division behavior of cells |
markers include tumor-associated antigens, |
||
secreted hormones, and enzymes. The table |
|||
|
|
||
The body’s cells are normally subject to strict |
lists a few examples. |
||
“social” control. They only divide until they |
The transition from a normal to a trans- |
||
come into contact with neighboring cells; cell |
formed state is a process involving several |
||
division then ceases due to contact inhibition. |
steps. |
||
Exceptions to this rule include embryonic |
1. Tumor initiation. Almosteverytumorbe- |
||
cells, cells of the intestinal epithelium (where |
gins with damage to the DNA of an individual |
||
the cells are constantly being replaced), cells |
cell.Thegeneticdefectisalmostalwayscaused |
||
in the bone marrow (where formation of |
by environmental factors. These can include |
||
blood cells takes place), and tumor cells. Un- |
tumor-inducing chemicals (carcinogens—e.g., |
||
controlled cell proliferation is an important |
components of tar from tobacco), physical |
||
indicator of the presence of a tumor. While |
processes (e.g., UV light, X-ray radiation; see |
||
normal cells in cell culture only divide 20–60 |
p.256), or in rare cases tumor viruses (see |
||
times, tumor cells are potentially immortal |
p. 398). Most of the approximately 1014 cells |
||
and are not subject to contact inhibition. |
in the human body probably suffer this type |
||
In medicine, a distinction is made between |
of DNA damage during the average lifespan, |
||
benign and malignant tumors. Benign tumors |
but it is usually repaired again (see p. 256). It |
||
consist of slowly growing, largely differenti- |
is mainly defects in proto-oncogenes (see |
||
ated cells. By contrast, malignant tumors |
p. 398) that are relevant to tumor initiation; |
||
show rapid, invasive growth and tend to |
these are the decisive cause of transformation. |
||
form metastases (dissemination of daughter |
Loss of an anti-oncogene (a tumor-suppressor |
||
lesions). The approximately 100 different |
gene) can also contribute to tumor initiation. |
||
types of tumor that exist are responsible for |
2. Tumor promotion is preferential prolif- |
||
more than 20% of deaths in Europe and North |
eration ofa cell damaged by transformation. It |
||
America. |
|
is a very slow process that can take many |
|
|
|
years. Certain substances are able to strongly |
|
B. Transformation |
accelerate it—e.g., phorbol esters. These occur |
||
in plants (e.g., Euphorbia species) and act as |
|||
|
|
||
The transition of a normal cell into a tumor |
activators of protein kinase C (see p. 386). |
||
cell is referred to as transformation. |
3. Tumor progression finally leads to a |
||
Normal cells have all the characteristics of |
macroscopically visible tumor as a result of |
||
fully differentiated cells specialized for a par- |
growth. When solid tumors of this type ex- |
||
ticular function. Their division is inhibited |
ceed a certain size, they form their own vas- |
||
and they are usually in the G0 phase of the |
cular network that supplies them with blood |
||
cell cycle (see p. 394). Their external shape is |
(angiogenesis). Collagenases (matrix metallo- |
||
variable and is determined by a strongly |
proteinases, MMPs) play a special role in the |
||
structured cytoskeleton. |
metastatic process, by loosening surrounding |
||
In contrast, tumor cells divide without in- |
connective tissue and thereby allowing tumor |
||
hibition and are often de-differentiated—i.e., |
cells to disseminate and enter the blood- |
||
they have acquired some of the properties of |
stream. Newapproaches to combating tumors |
||
embryonic cells. The surface of these cells is |
have been aimed at influencing tumor angio- |
||
altered, and this is particularly evident in a |
genesis and metastatic processes. |
||
disturbance of contact inhibition by neighbor- |
|
||
ing cells. The cytoskeleton of tumor cells is also restructured and often reduced, giving them a rounded shape. The nuclei of tumor cells can be atypical in terms of shape, number, and size.
Tumor markers are clinically important for detecting certain tumors. These are proteins that are formed with increasing frequency by
|
Cell proliferation |
401 |
A. Division behavior of cells |
|
|
Growth inhibition |
Uncontrolled |
|
due to contacts |
cell proliferation |
|
with adjacent cells |
|
|
|
Nutrient |
|
|
medium |
|
Normal cells |
Tumor cells |
|
B. Transformation
|
|
|
|
|
Normal cell |
Indicators: |
|
|
|
|
|
|
|
|
|
Tumor initiators |
|
|
|
|
|
Differentiated |
|
|
|
|
|
|
Non-dividing |
||
|
|
H3C |
|
|
|
|
|
|
|
|
|
|
|
Defined form |
|
|
|
|
|
|
|
|
|
Viruses |
|
|
|
|
|
|
|
Carcinogenic |
|
|
|
|
|
|
|
chemicals |
|
|
|
|
1 |
Tumor initiation: |
|
|
|
|
|
|
Genetic damage |
||
Physical processes |
UV |
|
|
|
|||
|
|
|
|
|
|||
|
|
|
|
|
|
||
Tumor promoters |
OH |
OH |
|
2 |
Tumor progression: |
||
|
|
H3C |
CH3 |
||||
e.g. Esters |
|
|
Preferential propagation |
||||
|
H |
|
|
|
|||
|
|
CH3 |
|
|
|
||
of phorbol |
H3C |
OH |
H |
|
|
|
|
|
|
|
|
|
|
||
Hormones |
O |
HO |
|
|
|
|
Indicators: |
|
|
|
|
De-differentiated |
|||
Phorbol |
CH2OH |
|
Tumor cell |
||||
|
|
|
|
Uncontrolled cell division |
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Altered cell surface |
Tumor markers (examples) |
|
|
|
|
Altered cytoskeleton |
||
|
|
|
|
and nucleus |
|||
Tumor-associated antigens 
CEA |
Carcinoembryonic |
|
Tumor progression: |
|
antigen |
3 |
|
|
α1-Fetoprotein |
Acquisition of malignancy |
|
AFP |
|
||
|
|
||
Hormones |
Calcitonin |
|
|
|
ACTH |
|
|
Enzymes |
Acid phosphatase |
|
|
Cell proliferation |
403 |
A. Alkylating agents, anthracyclines
Cross-linking |
“Bending” |
of DNA |
of the DNA |
components |
double helix |
|
|
H |
|
|
|
|
|
|
O |
|
OH |
|
O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
C |
|
|
|
|
|
|
|
|
|
|
|
N |
CH2 |
CH2 |
Cl |
|
|
|
|
|
|
CH2OH |
|
|
|
H3N |
Cl |
||||
|
O |
P |
|
|
|
|
|
|
OH |
|
|
|
|
Pt |
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
CH2 |
CH2 |
Cl |
|
|
|
|
|
|
|
|
|
|
|
H3N |
Cl |
|||
|
|
|
|
|
|
|
H CO |
|
O |
|
OH H |
O |
|
|
|
|
|
|
|
|
|
|
Cyclophosphamide |
3 |
|
Adriamycin |
|
|
|
|
|
|
Cisplatin |
||||||||||
|
|
|
R |
|
|
|
|
|
|||||||||||||
B. Antimetabolites |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
S |
CH3 |
|
|
|
|
SH |
|
|
|
|
|
|
|
|
SH |
|
|
||
|
N |
C |
|
N |
|
|
|
N |
C |
C |
N |
|
|
|
|
|
|
N |
C |
C |
N |
|
C |
CH |
2 |
|
|
|
|
1 |
|
|
|
|
|||||||||
|
HC |
C |
|
|
HC |
|
C |
CH |
|
|
|
|
HC |
|
C |
CH |
|||||
|
N |
|
|
|
N |
N |
|
|
|
|
|
N |
N |
||||||||
|
|
N |
|
|
|
SAH |
SAM |
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
P |
|
Rib |
|
|
P |
|
Rib |
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
me-tIMP |
|
|
|
|
|
|
|
|
tIMP |
|
|
|
|
|
6-Mercaptopurine |
||||||
|
|
GIn |
|
|
Glu |
+ P P |
|
|
|
tIMP |
tGMP |
tGDP |
|
|
tdGDP |
|
|||||
|
|
|
|
|
|
Phospho- |
Purine |
|
|
|
|
|
|
|
|
|
|
|
|
|
DNA |
|
|
|
|
|
|
synthesis |
|
|
|
|
|
|
|
|
|
|
|
|
|
||
PRPP |
|
3 |
|
ribosyl |
|
IMP |
GMP |
GDP |
|
4 |
|
dGDP |
|
||||||||
|
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
amine |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Hypoxanthine phospho- |
|
|
Amidophosphoribosyl |
|
|
|
|
O |
|
|
||||||||||
1 |
|
3 |
|
|
|
|
C |
|
OH |
||||||||||||
ribosyltransferase 2.4.2.8 |
|
transferase 2.4.2.14 |
|
|
|
|
|
||||||||||||||
|
|
|
|
|
H2N |
N |
|||||||||||||||
2 |
Thiopurine methyl- |
|
4 |
Ribonucleoside diphosphate |
|
|
|
||||||||||||||
|
|
|
|
|
H |
|
|||||||||||||||
transferase2.1.1.67 |
|
reductase 1.17.4.1 |
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Hydroxyurea |
||||
|
|
O |
|
|
Precursors |
|
|
|
|
|
|
H2N |
|
N |
|
N |
|
|
|
||
|
|
C |
|
|
F |
|
N5,N10-methylene-THF |
|
C |
C |
CH |
|
|
||||||||
|
|
|
C |
|
|
|
|
|
|
|
|
|
N |
|
C |
|
C |
|
|
||
|
HN |
|
|
dUMP |
H C |
H |
|
|
|
|
|
|
C |
N |
CH2 |
||||||
|
|
C |
|
CH |
|
|
|
|
N |
A |
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
O |
N |
|
|
|
THF |
|
|
|
|
|
NH2 |
H3C |
N |
|
||||||
|
|
|
|
|
|
|
|
P |
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
P |
Rib |
|
|
5 |
|
|
|
|
6 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
5-fluoro- |
|
|
|
|
|
|
|
|
|
N |
A |
Methotrexate |
|
|
|
||||||
deoxyuridine |
|
|
DHF |
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
P |
(amethopterin) |
|
|
|
|||||||||||
monophosphate |
|
|
|
|
|
|
C |
O |
|||||||||||||
dTMP |
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
Dihydrofolate |
|
|
|
|
|
|
|
|
HN |
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
OOC |
CH2 |
CH2 |
C |
COO |
|||||||
|
|
|
|
|
|
dTTP |
|
|
|
|
|
|
|
||||||||
5-fluorouracil |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
5-fluoro- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
deoxyuridine |
|
DNA |
|
|
|
|
|
|
5 |
Thymidylate synthase 2.1.1.45 |
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
6 |
Dihydrofolate reductase 1.5.1.3 |
|||||||