



|
|
|
|
|
|
|
|
|
|
|
|
|
|
Genetic engineering |
259 |
|
A. Restriction endonucleases |
|
|
|
|
|
|
|
|
|
|||||||
|
H |
H |
|
|
|
|
|
|
|
|
|
Palindrome |
|
|
|
|
|
H 3' |
|
H G |
|
|
5' |
G |
A |
A |
T |
T |
C |
3' |
|
|
|
|
O |
H |
|
|
|
|
3' |
C |
T |
T |
A |
A |
G |
5' |
|
|
|
|
|
|
H2C5' |
|
|
|
|||||||||
O |
P |
O |
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
O |
|
|
|
H |
|
|
|
EcoRI |
|
DNA-Ligase |
|
|
|
||
|
A |
|
|
|
3.1.21.4 |
|
6.5.1.1 |
|
|
|
||||||
|
|
H |
3' |
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
5' |
G |
A |
A |
T |
T |
C |
3' |
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
H 3' |
|
H G |
|
|
3' |
C |
T |
T |
A |
A |
G |
5' |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
OH H |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
5' |
|
A |
A |
T |
T |
C |
3' |
|
|
O |
P |
O |
|
H2C |
5' |
|
G |
|
|
|
|
G |
|
|
|
|
|
O |
|
|
|
|
|
5' |
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|||||||
A |
|
3' |
C |
T |
T |
A |
A |
|
|
|
||||||
|
O |
|
|
H |
|
EcoRI |
+ DNA |
|
||||||||
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
3' |
|
|
Overhanging ends |
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
B. DNA cloning |
|
|
|
|
|
|
|
|
DNA |
G A A T T C |
G A A T T C |
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
C T T A A G |
C T T A A G |
|
|
|
|
|
|
EcoRI |
|
|
EcoRI |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Purification |
|
|
|
|
Vector DNA |
|||
|
|
|
|
|
|
(Plasmid) |
||
Isolated |
DNA ligase |
|
|
|
|
|
|
|
DNA fragment |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
A |
|
|
|
|
|
|
|
|
A |
|
|
|
|
|
|
|
|
T |
|
|
Host cell |
|
Gene for |
|
|
|
|
T |
C |
(bacterium) |
|
|
|
|
|
|
|
|
|
antibiotic |
G |
|
|
|
|
|
|
|
|
resistance |
|
A |
A |
|
G |
|
|
|
|
|
|
||||
|
|
|
T |
|
|
|||
|
|
|
T |
|
|
|
|
|
|
|
|
C |
|
|
|
|
|
|
|
Recombinant |
|
|
|
|
|
|
|
|
plasmid |
|
|
|
|
|
|
Transformation |
|
|
|
|
|
|
|
|
|
Plasmid |
|
Plasmid |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
isolation |
|
|
|
|
|
Bacterial |
|
|
Cleavage |
|
|
|
|
|
|
|
with |
|
|
|
|
|
|
genome |
|
|
|
|
|
|
|
|
|
|
EcoRI |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Bacterial culture in the |
|
|
|
Cloned |
|
|
|
|
presence of antibiotic |
|
|
|
DNA fragment |
|||


|
|
|
Genetic engineering |
261 |
|
A. Gene libraries |
|
|
|
|
|
|
|
|
Imprint on |
|
|
|
Gene library |
Host cell |
plastic membrane |
|
|
|
|
|
|
|
|
|
in phages |
|
|
Gene probe |
|
|
|
|
|
||
|
Plate |
|
|
Hybrid- |
|
|
with phage plaques |
|
|
||
|
|
ization |
|
||
|
|
Positive plaque |
|
|
|
|
|
|
|
|
|
|
|
|
|
Washing, |
|
|
|
|
|
detection |
|
|
|
|
|
of the |
|
|
|
|
|
gene probe |
|
|
DNA |
|
Gene |
Gene probe |
|
|
isolation |
|
|
||
|
|
|
|
|
|
|
Restriction |
Phage |
|
|
|
Cloned gene |
repli- |
|
|
|
|
|
|
|
|
||
|
cation |
|
Membrane |
|
|
|
|
|
|
||
B. Sequencing of DNA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
G A T C |
a |
5' |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3' |
||
|
3' |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5' |
Protein |
|
|
Synthetic primer |
|
Single-stranded preparation |
|
sequence |
||||||||||||||||
|
|
|
||||||||||||||||||||
|
|
|
DNA |
|||||||||||||||||||
|
5' |
|
|
|
|
|
|
|
|
|
|
Hybridization |
||||||||||
b |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
sequence |
|
|
3' |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3' |
N |
|
|
|
|
|
|
|
|
C |
|
c |
|
|
|
|
|
|
|
T |
Phe |
|
|
|
|
|
|
|
|
T |
|
|
G |
A |
T |
|
|
C |
|
C |
|
|
|
|
|
|
|
|
|
A |
Tyr |
+ 4 dNTP |
+ 4 dNTP |
+ 4 dNTP |
|
+ 4 dNTP |
T |
|
|||
+ ddGTP |
+ ddATP |
+ ddTTP |
|
+ ddCTP |
T |
|
|||
+ Polymerase |
+ Polymerase |
+ Polymerase |
|
+ Polymerase |
C |
Ala |
|||
|
|
|
|
|
|
|
|
G |
|
|
|
|
|
|
|
|
|
T |
|
|
|
|
Example: G |
G |
A |
T |
C |
C |
Thr |
|
|
|
|
|
|
|
A |
|
|
|
|
|
= ddG |
|
|
|
|
|
|
|
|
|
|
|
|
|
A |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
A |
Glu |
|
|
|
|
|
|
|
|
G |
|
|
|
|
|
|
|
|
|
A |
|
|
|
|
|
|
|
|
|
A |
Glu |
|
|
|
|
|
|
|
|
G |
|
d |
Gel electrophoresis |
|
|
|
|
|
G |
|
|
|
|
|
|
|
T |
Met |
|||
e |
Visualization of the fragments |
|
|
|
|
|
|||
|
|
|
|
|
A |
|
|||
f |
Reading off ( |
) : ACGATG ... |
|
|
|
|
|
|
|
|
|
|
|
|
5' |
N |
|||
|
|
|
|
|
|
|
|
||
1. Method |
2. Sequence pattern |
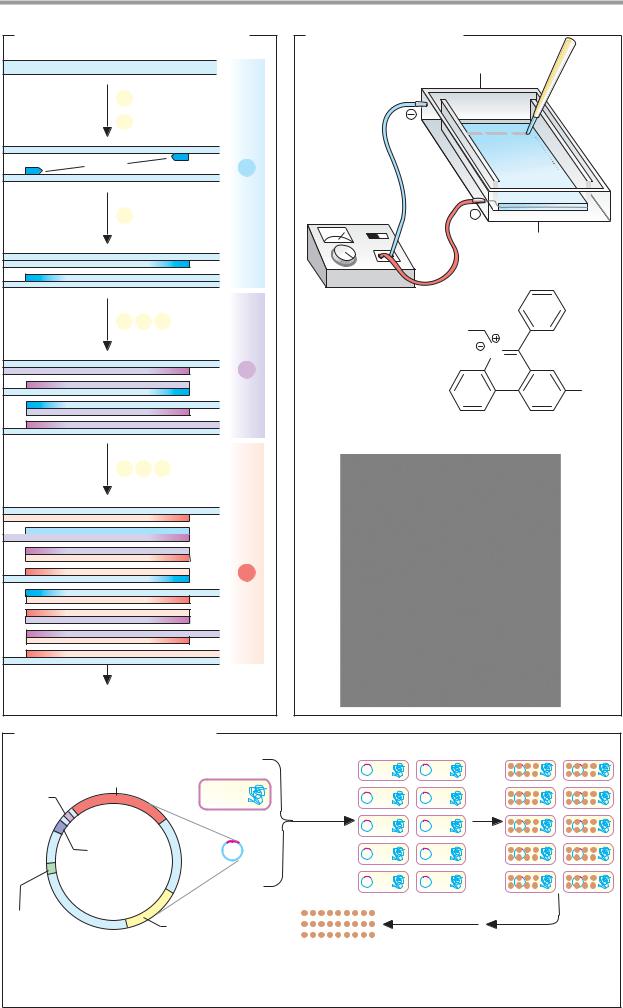
|
|
|
|
|
Genetic engineering |
263 |
|
A. Polymerase chain reaction (PCR) |
B. DNA electrophoresis |
|
|
||||
|
|
|
DNA |
Electrophoresis chamber |
Sample |
|
|
|
|
|
|
|
|
|
|
|
a |
Heat |
|
|
|
|
|
|
b Hybridize |
|
|
|
|
||
|
Primer |
|
Cycle |
|
|
|
|
|
|
1 |
|
|
|
|
|
|
|
De-novo synthesis |
Power supply |
|
|
|
|
|
c |
unit |
|
|
|
||
|
(DNA polymerase) |
|
|
|
|
||
|
|
|
|
|
|
Agarose gel |
|
|
a |
b |
c |
|
H3C |
|
|
|
|
|
|
|
|
|
|
|
|
|
Cycle |
|
Br |
N |
|
|
|
|
2 |
|
|
|
|
|
|
|
|
Ethidium bromide |
|
NH2 |
|
|
|
|
|
|
|
||
|
|
|
|
1 |
2 |
3 |
|
|
a |
b |
c |
|
|
|
bp |
|
|
|
|
|
|||
|
|
|
|
b |
|
|
1857 |
|
|
|
Cycle |
|
|
|
1058 |
|
|
|
3 |
|
|
|
|
|
|
|
|
|
|
929 |
|
|
|
|
|
a |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
383 |
|
etc |
|
|
|
|
|
|
C. Overexpression of proteins |
|
|
|
|
|||
Ribosome |
Gene to be |
Host cell |
|
|
|
|
|
binding |
expressed |
|
|
|
|
|
|
site |
|
|
|
Trans- |
|
|
|
|
|
|
|
formation |
Induction |
|
|
|
Inducible |
|
Repli- |
|
|
||
|
|
cation |
|
|
|
||
|
promotor |
|
|
|
|
|
|
Origin of |
|
|
Gene for |
|
|
|
|
replication |
|
|
antibiotic |
|
|
|
|
|
|
|
resistance |
Overexpressed |
Protein |
Cell |
|
|
|
|
|
protein |
purification |
lysis |
|
Expression plasmid

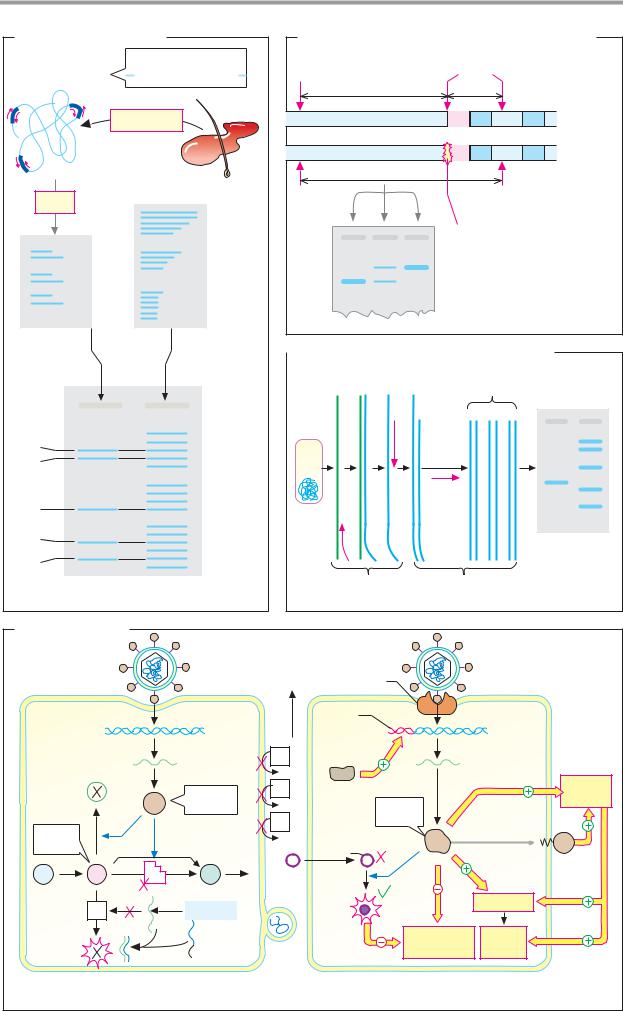
|
|
|
|
|
Genetic engineering |
265 |
||
A. DNA fingerprinting |
B. Diagnosis of sickle-cell anemia using RFLP |
|
||||||
|
STR, z.B. |
Mst II |
|
|
|
Mst II |
|
|
DNA |
TCTATCTGTCTG |
|
|
|
|
|
||
|
|
1200 bp |
|
201 bp |
|
|
||
|
|
|
|
|
|
|||
|
Extraction |
|
|
|
|
|
Normal |
|
|
|
|
|
|
|
gene |
|
|
|
|
|
|
|
E1 |
I1 |
|
|
|
|
|
|
|
E2 |
|
||
|
|
|
|
|
|
|
Sickle-cell |
|
|
|
|
|
1401 bp |
|
gene |
|
|
|
|
|
|
|
|
|
||
PCR |
Evidence |
Mst II |
|
|
|
Mst II |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Mutation |
|
|
|
Locus 1 |
1.4 |
|
|
|
|
|
|
|
Locus 2 |
|
|
|
1 Normal (A/A) |
|
||
|
1.2 |
|
|
|
|
|||
|
Locus 3 |
|
1 |
2 |
3 |
2 Heterozygotic (A/S) |
|
|
|
kbp |
3 Homozygotic (S/S) |
|
|||||
|
|
|
|
|
|
|||
Amplificate |
Comparative |
|
C. Evidence of viral DNA using RT-PCR |
|
fragments |
|
|
|
|
||
|
|
|
|
|
|
|
|
RNA Hy- |
ss- |
ds- |
|
|
|
|
|
|
|
|
brid |
DNA |
DNA |
Amplificate |
Standards |
|
Alleles |
|
|
|
|
|
P2 |
|
|
|
|
|
|
|
Virus |
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
||
1/3 |
|
|
Locus 1 |
|
|
|
PCR |
|
|
|
1/4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Locus 2 |
|
|
|
P3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2/4 |
|
|
|
|
P1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3/3 |
|
|
Locus 3 |
|
|
|
|
|
|
Agarose gel |
3/5 |
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
Agarose gel |
|
|
Reverse |
|
DNA polymerase |
|
|||
|
|
|
transcriptase |
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
||
D. Gene therapy |
|
|
|
|
|
|
|
|
|
|
|
|
|
Viral vector with |
Tumor- |
|
|
|
Viral vector |
|
|
|
|
|
specific |
|
|
|
with foreign DNA |
|||
|
|
|
foreign DNA |
|
|
|
|
|||
|
|
|
|
receptor |
|
|
|
|
||
|
|
|
|
|
|
|
|
|
||
Normal |
|
|
|
|
Tumor- |
|
|
|
Tumor cell |
|
body cell |
|
|
DNA |
|
specific |
|
|
|
DNA |
|
|
|
|
|
promoter |
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
Harmless |
|
|
mRNA |
E1 |
|
|
|
mRNA |
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
product |
|
|
Gene |
E1 |
|
|
|
|
|
Immune |
|
|
|
|
|
|
|
|
system |
||
|
|
|
|
|
Gene |
|
|
|||
|
|
|
product |
|
|
|
|
|
||
|
|
|
|
E1 |
|
product |
|
|
|
|
Accu- |
|
|
|
Cytostatic |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
mulates |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
agent |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
A |
B |
E1 |
C |
|
|
|
|
|
|
|
|
E2 |
|
Genome |
|
|
|
|
|
Apoptosis |
|
|
|
|
|
|
|
|
|
|
||
Toxic |
|
|
Antisense |
|
|
|
Cell |
|
Cell |
|
product |
|
|
DNA |
Lipo- |
|
|
proliferation |
death |
|
|
|
|
|
|
some |
|
|
|
|
|
|
1. For metabolic defects |
|
|
2. For tumors |
|
|
|
||||