

Введение
Предмет органической химии
Основным объектом изучения химии являются химические соединения и их превращения.
Ранее в курсах общей и неорганический химии уже состоялось знакомство с основополагающими и всеобщими законами химии, которые определяют само существование химических соединений и протекание реакций между ними. Изучались также свойства и превращения соединений, которые составлены из элементов, входящих в периодическую таблицу Д.И. Менделеева. Эти соединения были названы неорганическими.
Органическая химия – это обособленный раздел химической науки. Она изучает углеводороды и их производные, т. е. – соединения углерода с другими элементами, чаще всего с водородом, кислородом, азотом, серой, фосфором и галогенами. Такая приверженность одному элементу, углероду, обусловлена способностью углеродных атомов образовывать четыре ковалентные, а также кратные связи с атомами углерода и другими элементами, и склонностью соединяться в прямые и разветвленные цепи, которые могут включать от двух до нескольких тысяч атомов углерода. В настоящее время известно более 15 миллионов органических соединений, что больше чем на порядок превосходит число соединений всех других элементов. Но даже эта цифра не ограничивает возможности «конструирования» новых органических молекул.
Превращения органических соединений управляются общими законами химии, но имеются также специфические закономерности, характерные только для органических веществ. Органическая химия изучает более высокоорганизованную материю, чем неорганическая химия. Органические соединения появились во Вселенной позже неорганических, они являются носителями жизнедеятельности организма человека, животных, растений. Органические вещества составляют основу многих важнейших отраслей химической промышленности: нефтехимии, пластических масс, синтетического каучука, моторного топлива, смазочных материалов, лаков, красок, медикаментов, взрывчатых веществ, текстильных, кожевенных и пищевых материалов и других отраслей.
Некоторые природные органические соединения и полученные из них продукты люди использовали еще в древние времена. Например, спиртные напитки (вино, пиво, «мёд»), уксус, органические красители (пурпур, индиго, ализарин), эфирные масла, сахар (тростник) и другие. Первым методом переработки природных веществ была перегонка, которая позволила выделить и получить целый ряд индивидуальных веществ. После открытия во второй половине XVIII века Ломоносовым и Лавуазье закона сохранения вещества в химию вошёл химический анализ. Прогресс в области анализа способствовал совершенствованию методов очистки органических соединений. В первой половине XIX века органическая химия выделилась в самостоятельную дисциплину. Многие ученые тогда полагали, что органические вещества возникают только в живом организме под влиянием особой «жизненной силы». Этот взгляд на происхождение органических веществ получил название витализма. Однако в 1824 году немецкий химик Вёлер получил из неорганических веществ щавелевую кислоту, а в 1828 году – мочевину. Далее русский ученый Н.Н.Зинин синтезировал анилин (1842 г.), в 1845 г. немец Кольбе – жиры, в 1861г. А.М.Бутлеров – первое сахаристое вещество. Успешно развивающийся начиная с первой половины XIX века органический синтез навсегда покончил с витализмом.
Строение и реакционная способность органических соединений
Исторически первой системой взглядов в области органической химии явилась теория радикалов (Дюма, Либих, Берцелиус). Согласно этой теории многие превращения органических соединений протекают так, что остаток, состоящий из нескольких атомов, не изменяясь, переходит из одного органического соединения в другое. Эти группы атомов и были названы «радикалами». Однако вскоре было установлено, что атомы водорода в радикалах могут быть заменены на другие, например, атомы хлора. Таким образом, неизменность радикала нарушилась.
Теория типов, высказанная в 1853 г. французским химиком Ш. Жераром, рассматривала изменения, которые претерпевают органические соединения. В согласии с этой теорией органические соединения реагируют подобно простым неорганическим веществам, которые являются типами. Так к типу водорода относятся углеводороды, к типу хлористого водорода – галогенпроизводные, к типу воды – спирты, кислоты, эфиры и т.д.
Теория типов, хотя и с трудом, позволила классифицировать большой фактический материал, накопленный органической химией к этому времени. Но она не могла помочь в познании строения и путей синтеза органических соединений.
Важными вехами в развитии теоретических представлений в органической химии явилось установление четырехвалентности углерода немецкими учеными Кекуле и Кольбе (1857 г.) и способности углерода образовывать цепочки атомов (Кекуле и Купер, 1857 г.) Осново-полагающей явилась теория химического строения органических соединений Александра Михайловича Бутлерова (1861г.). Сущность этой теории заключается в трех основных положениях:
все химические вещества имеют строго определенное химическое строение, то есть строго определенный порядок чередования атомов в молекуле, определенную закономерность во влиянии атомов друг на друга;
химическое строение веществ определяет их физические и химические свойства;
изучение свойств веществ позволяет определить их химическое строение.
Теория химического строения позволила классифицировать весь накопившийся экспериментальный материал, предсказала возможное число органических соединений одного определенного состава (изомерия) и пути их синтеза. В XX веке она получила обоснование с позиций квантовой механики. Позднее (1877 г.) теория строения была дополнена Вант-Гоффом и Ле Белем теорией пространственного расположения атомов в молекулах – стереохимической теорией.
Создание теории химического строения способствовало бурному развитию органической химии и в последней четверти XIX века она приняла современный вид.
Прежде чем переходить к рассмотрению свойств органических соединений определенных классов, необходимо рассмотреть несколько общих вопросов, касающихся строения, методов исследования и классификации органических соединений.
Согласно теории химического строения каждое вещество должно иметь одно определенное химическое строение. В то же время одной молекулярной формуле может соответствовать несколько веществ – столько, сколько можно построить из данного количества атомов, учитывая правила валентности.
Вещества, тождественные по составу, но различающиеся по химическому строению, называются изомерами. Примеры изомеров С4Н10 и С4Н10О:
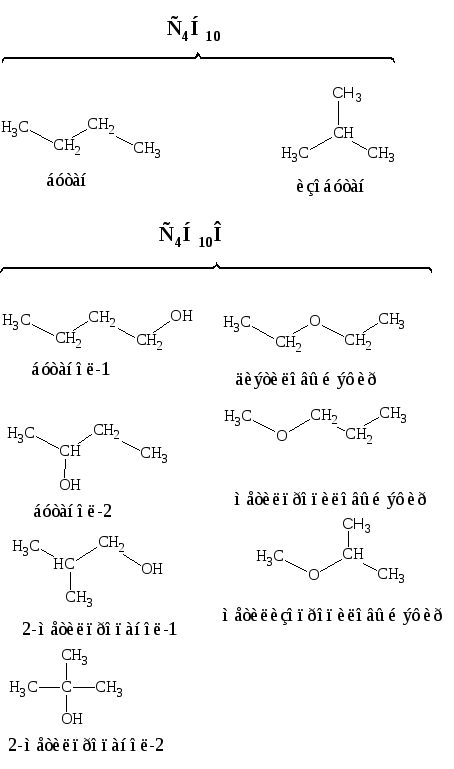
Изомеры различаются по своим свойствам и могут принадлежать к различным классам органических соединений.
Химические свойства органических соединений зависят от природы составляющих её атомов и от их взаимного расположения. Естественно, атомы в молекулах меняют свое состояние и влияют друг на друга. Наиболее сильно взаимодействуют, конечно, атомы, образующие непосредственную связь между собой. Но и атомы, не связанные непосредственно, взаимно влияют друг на друга через другие атомы по цепочке (поляризация) или через окружающее пространство (эффект поля). Взаимное влияние непосредственно связанных между собой атомов зависит, в первую очередь, от их природы и характера связи между ними. Различают два основных вида связей – ковалентную и электровалентную, или ионную. Ковалентная связь возникает при обобщении неспаренных валентных электронов с разными спинами, электровалентная – при передаче неспаренного электрона одного атома другому с образованием ионов, которые взаимно притягиваются.
А+В→ АВ А+В→А++В
Одним из вариантов ковалентной связи является координационная, когда атомы объединяют пару электронов одного из них.
Разновидностью координационной связи является связь семиполярная. В этом случае атом-донор заряжается положительно, атом-акцептор – отрицательно:
А+В→ А+ В (А→В)
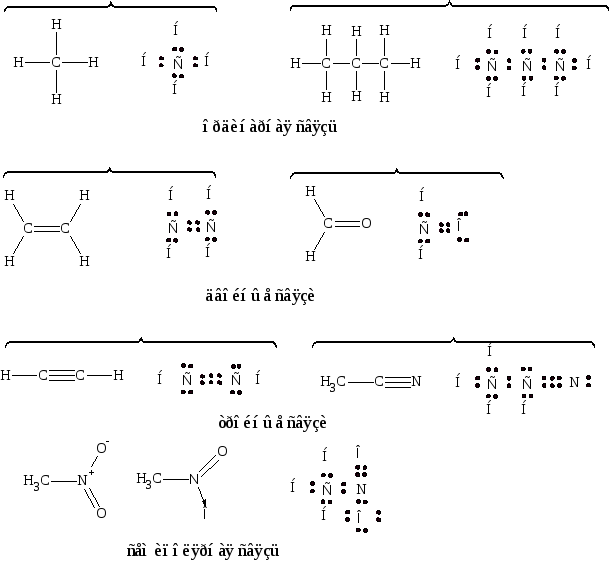
В органических соединениях встречается преимущественно ковалентная связь. Поскольку углерод четырехвалентен, для каждого органического соединения можно записать так называемую октетную формулу. В этих формулах учитываются только внешние валентные электроны как участвующие, так и не участвующие в связи.
Черточка при написании структурной формулы заменяет пару электронов (Купер). Октетными формулами мы будем пользоваться в дальнейшем. Они помогают понять строение и химизм реакций органических соединений.
Валентное состояние атомов, как известно, определяется распределением электронов на внешних орбитах. Органическую химию интересуют, главным образом, семь низших орбит: 1s, 2s, 2p, 3s, 3p, 3d, 4s.
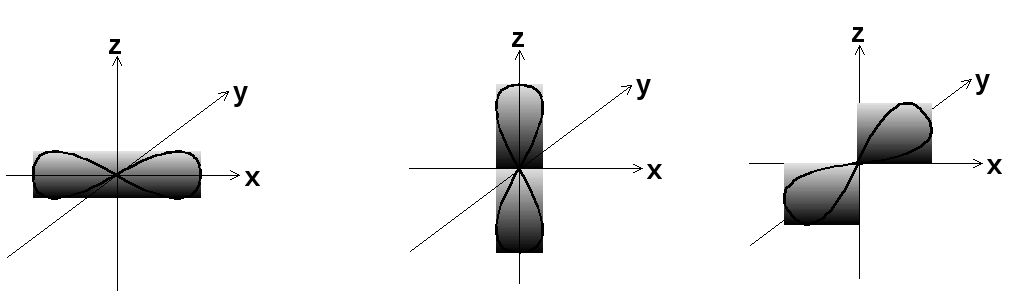
Вероятность нахождения электрона в определенном месте пространства на s-орбитах не зависит от направления от ядра, т.е. эти орбиты шаровые.

Для электронов в р-состоянии в атоме имеются три одинаковые взаимно перпендикулярные орбиты, причем электронные облака по каждому направлению имеют форму «объёмных восьмёрок», или «гантелей».
В соответствии с правилами квантовой механики состояния электронов на атомных орбитах для некоторых элементов можно представить следующим образом:
-
Н (1)
↑
1s
↑↓
↑
↑
2s22p2
С(6)
↑↓
1s2
↑↓
↑
↑
↑
2s22p3
N (7)
↑↓
1s2
↑↓
↑↓
↑
↑
2s22p4
O (8)
↑↓
1s2
↑↓
↑↓
↑↓
↑
2s22p5
F (9)
↑↓
1s2
Судя по числу неспаренных электронов, углерод должен иметь валентность 2 , азот – 3, кислород – 2 , фтор – 1. Действительно, эти элементы проявляют соответствующие валентности.
Простая ковалентная связь между двумя атомами возникает при переходе двух электронов на общую для двух ядер орбиту. Орбиты этого типа называются молекулярными. При образовании, например, молекулы водорода атомы водорода сближаются, их электронные облака 1s-электронов перекрываются и две атомные орбиты заменяются молекулярной, которая имеет яйцевидную форму и называется -орбитой.

Химическая связь, образованная -орбитой, называется -связью.
При перекрывании р-орбиты хлора с s-орбитой водорода также образуется ковалентная -связь.
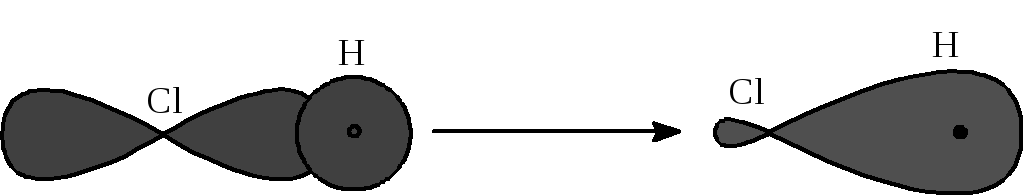
В изолированном атоме углерода L-оболочка имеет строение 2s22pх2py, и углерод должен проявлять валентность 2. Однако если с затратой определенного количества энергии один из 2s электронов поднять на свободную 2рz-орбиту, углеродный атом приобретет структуру 1s2 2s 2pх 2py 2рz и будет иметь 4 неспаренных электрона: три – 2р и один – 2s.
-
↑↓
↑
↑
2
 s22p2
s22p2
С(6)
↑↓
1s2
-

↑
↑
↑
↑
2s 2pх 2py 2рz
С(6)
↑↓
1s2
При этом атом углерода сможет образовать 4 связи, например, с атомами водорода. Но три р-орбиты перекрываются s-орбитами водорода эффективно, а четвертая 2s-орбита из-за пространственных затруднений перекрывается неполно. В результате должны образоваться три прочные С–Н связи и одна слабая. В действительности, все четыре С–Н связи в метане (СН4) равноценны. Причина в следующем. В связанном углеродном атоме электроны распределяются не на «чистых» s- и р-орбитах, а на смешанных, гибридных (теория гибридизации Полинга). Гибридные орбиты имеют более высокую энергию, чем s- и р-орбиты, но для образования связей они выгоднее геометрически, потому что полнее перекрываются орбитами других атомов. Связи при этом получаются прочными. Таким образом, у атома углерода в метане 4 атомные орбиты 2s 2pх 2py 2рz заменяются четырьмя идентичными гибридными орбитами в sp3 валентном состоянии. Эти орбиты имеют одинаковый вид и энергию. Они направлены вдоль некоторых осей к вершинам правильного тетраэдра (тетраэдрическое строение).
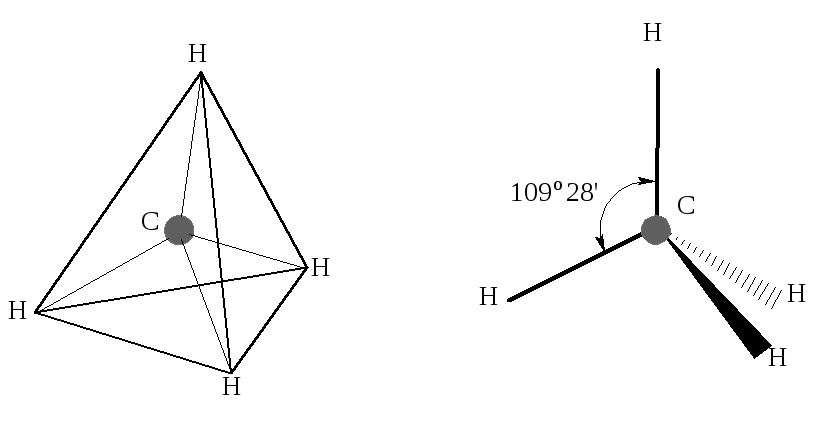
Атом углерода в молекуле метана и других насыщенных углеводородах находится в состоянии sp3-гибридизации.
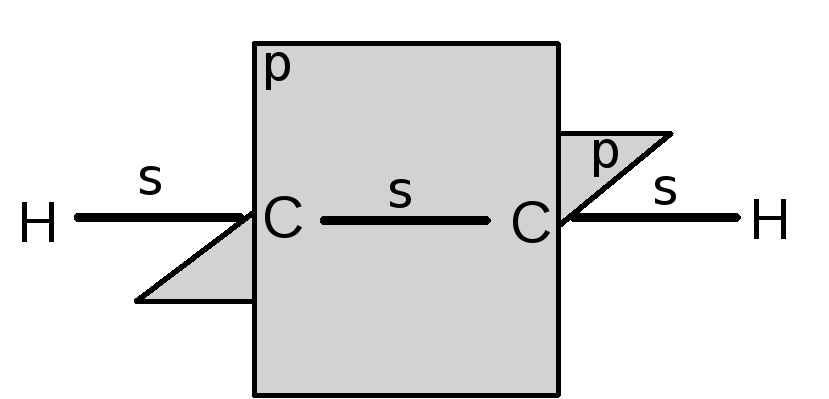
Кроме тетраэдрической sp3-гибридизации для углерода возможны также иные виды гибридизации. Например, при образовании двойных связей в ненасыщенных соединениях гибридизации подвергаются одна s- и две р-орбиты. Углерод при этом находится в sp2-гибридном состоянии. При этом оси всех трех орбит лежат в одной плоскости. Оставшаяся негибридизованной третья р-орбита располагается по оси перпендикулярно этой плоскости. Каждый атом углерода образовывает 3 -связи, одна из них связывает атомы углерода. Кроме того, на периферии молекулы боковыми поверхностями перекрываются р-орбиты углеродных атомов с параллельными осями.
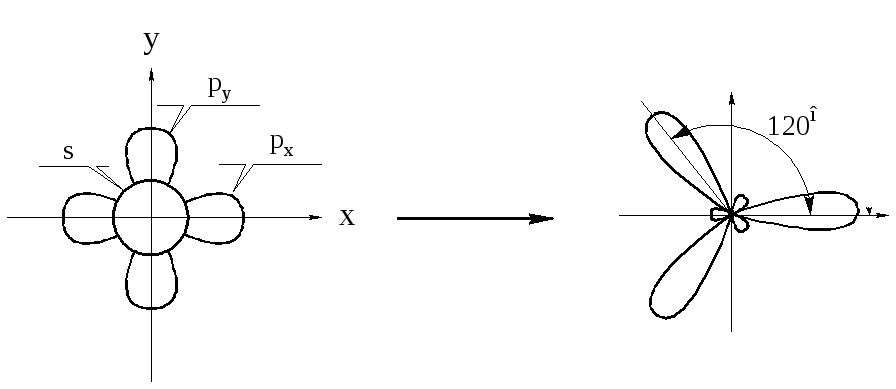
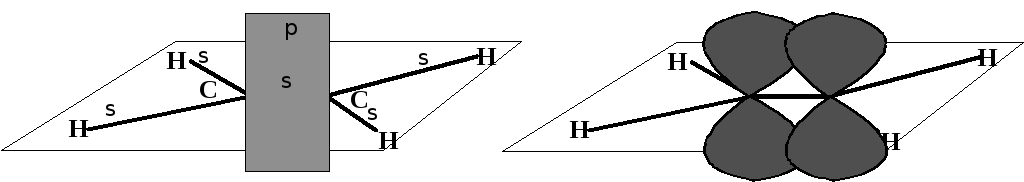
Такое взаимодействие между атомами называется -связью (взаимодействие р-орбит). -Связь менее прочна, чем -связь, потому что р-облака перекрываются в малой степени. Прочность (+) связей в этилене 606,7 кДж∙моль-1. Прочность -связи – 350,0 кДж∙моль-1, отсюда -связь имеет энергию диссоциации 256,7 кДж∙моль-1.
При образовании тройной связи (например, в ацетилене) углеродные атомы находятся в sp-гибридном состоянии. Гибридная sp-орбита возникает комбинированием одного s- и одного р-электрона.
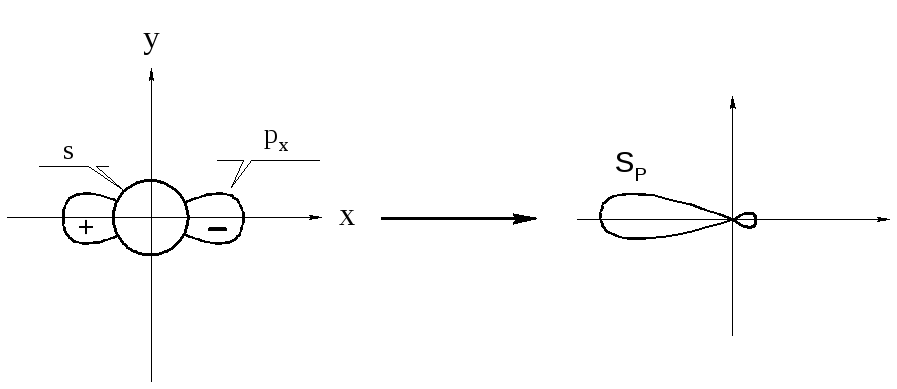
Две другие р-орбиты направлены перпендикулярно оси sp-орбиты и друг другу. Перекрывание sp-орбит углеродных атомов создает -связь, а перекрывание двух пар р-орбит – две -связи (показано условно).
Атомы углерода и водорода в ацетилене лежат на одной прямой, и молекула является линейной. Вторая -связь в ацетилене еще слабее, чем первая. Энергия её диссоциации – 215,5 кДж∙моль-1.
Рассмотренные нами современные представления о природе химической связи позволяют объяснить некоторые эффекты взаимного влияния атомов в молекуле: индукционный эффект, эффекты сопряжения и сверхсопряжения и некоторые другие. Электронные облака могут смещаться в молекуле так, что под их влияние попадают соседние атомы. Кроме того, поскольку электронные облака не имеют границ, их влияние может передаваться через пространство. Рассмотрим молекулу хлористого метила. Хлор как более электроотрицательный атом имеет большее сродство к электрону, чем углерод. Поэтому их общая электронная пара смещается в сторону атома хлора и он приобретает частичный отрицательный заряд. Углерод же получает частичный положительный заряд.
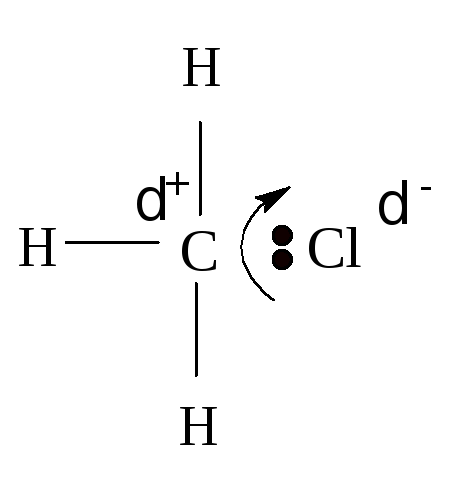
Образуется электрический диполь, который поляризует в соседних С–Н связях. Такой эффект называется индукционным.
В молекулах с несколькими двойными связями строение и свойства электронной оболочки сильно зависят от взаимного расположения двойных связей. Например, в молекуле бутадиена-1,3 р-электронные орбиты взаимно перекрываются.
СН2=СН–СН=СН2

Образуется единая система электронов, которая охватывает все четыре атома углерода. Этот эффект называется эффектом сопряжения. Сопряженные системы нельзя рассматривать как системы с изолированными независимыми двойными связями. Образование системы сопряжения приводит к выравниванию электронной плотности в сопряженном фрагменте и, как следствие, к энергетической стабилизации молекулы.
Эффект сверхсопряжения (гиперконъюгации) наблюдается в случае, когда насыщенная группа, например, метильная, связана с ненасыщенной группой. Вследствие смещения электронных облаков в сторону ненасыщенной группы атомы водорода в насыщенной группе активизируются, связь С–Н ослабляется. В свою очередь, ненасыщенная группировка поляризуется, ее атомы получают заряды.
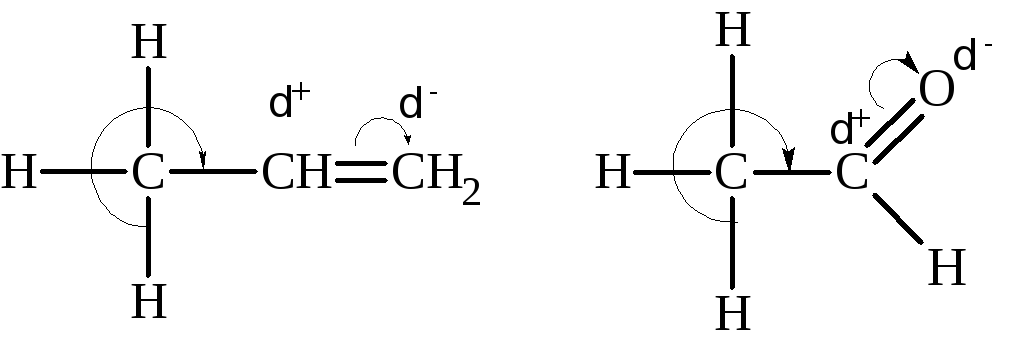
Определение строения органических соединений
Исследование органического вещества предполагает следующие последовательные стадии:
– очистка обычно осуществляется перегонкой, перекристал-лизацией, хроматографированием, экстракцией или другими методами.
– качественный и количественный анализ. Зная качественный (элементный) состав вещества, содержание в нём отдельных элементов и его молекулярную массу, можно определить молекулярную формулу соединения.
– определение и доказательство строения. Химическое строение органического вещества определяется как химическими, так и физическими методами.
Определение строения химическими методами заключается в изучении химических свойств исследуемого соединения путем проведения качественных реакций на наличие в нем различных функциональных групп. Знание химических свойств позволяет отнести вещество к тому или иному гомологическому ряду и определить некоторые особенности его структуры. Строение вещества подтверждается также встречным синтезом, т. е. синтезом искомого соединения другими путями и из других исходных веществ. Химические методы в настоящее время, как правило, используются в сочетании с исследованием вещества физическими методами.
Особенно мощным средством исследования строения органических веществ являются электромагнитные колебания. Дифракция рентгеновских лучей (длина волны = 0,1 – 0,0110-8 см) в кристаллах используется для определения параметров кристаллической решетки. Электромагнитные колебания в ультрафиолетовой ( = 10-6 – 410-6 см), видимой ( = 410-4 – 710-4 см) и инфракрасной ( = 710-4 – 10-2 см) областях спектра помогают определить тонкое строение молекул. Микроволновые колебания ( = 10-2 – 10 см) характеризуют вращение молекул. Сантиметровые и метровые волны с одновременным воздействием электрического поля используются для определения строения. Это методы электронного парамагнитного резонанса и ядерный магнитный резонанс. Чаще других используются инфракрасные (ИК) спектры, спектры комбинационного рассеяния (КР) и ядерного магнитного резонанса (ЯМР). Кратко рассмотрим сущность этих методов.
Спектры ИК и КР
Если вещество поместить в кювету из бромида калия и направить на него инфракрасные лучи, часть энергии излучения поглотится. Эта энергия затрачивается, в основном, на возбуждение колебаний атомов, то есть на изменение в молекулах длин связей и углов между ними. Вещество поглощает кванты вполне определенной энергии, иначе говоря – излучение определенной частоты. Если пучок ИК-излучения развернуть при помощи щели и призмы (например, из хлорида натрия) по частоте, получается ИК-спектр вещества. Поглощению энергии будут соответствовать на спектре минимумы. По спектру можно определить энергию (частоту) поглощенного света. Некоторые минимумы настолько характерны для отдельных групп атомов и связей, что положение их в спектре мало меняется при переходе от одной молекулы к другой. Колебания таких групп атомов и связей называются характеристическими. Они указывают на наличие или отсутствие в молекуле тех или иных групп атомов или связей. По отклонениям от характеристических колебаний судят об особенностях строения молекулы. Например, характеристические валентные колебаний С=О – 1700 см-1; С=N 2250 см-1; О–Н – 3550-3650 см-1 и т.д. Спектры комбинационного рассеяния, или рамановские спектры, тоже являются колебательными спектрами. Для их изучения измеряют спектр излучения, рассеянного веществом. Оба колебательных спектра дополняют друг друга.
Спектры ЯМР
Ядра элементов, имеющие магнитный момент (1Н, 19F, 13C, 31P и др.), в магнитном поле поглощают радиочастотное излучение. Поглощаемая энергия расходуется на изменение ориентации спинов ядер в магнитном поле. Магнитно-симметричные ядра (12С, 16О, 14N и др.) не поглощают энергию излучения и не проявляются в спектре ЯМР. Для исследования с помощью спектров ЯМР чаще всего используются протоны, т. к. они присутствуют почти в каждой органической молекуле. Метод в этом случае называется протонным магнитным резонансом (ПМР).
Для получения спектра ЯМР ампулу с исследуемым веществом помещают в однородное постоянное магнитное поле. Ампула окружена катушкой, в которой пропускают переменный ток для создания радиочастотного электромагнитного поля. Далее либо изменяют частоту электромагнитного поля при постоянном магнитном, либо плавно изменяют напряженность магнитного поля при неизменной частоте. Поглощение энергии фиксируется самописцем в виде пиков. Сигналы ЯМР для отдельных изотопов характеристичны. Однако атомные ядра в молекулах окружены электронами, которые экранируют их от действия магнитного поля. Характеристичность при этом несколько нарушается. Эти нарушения характеристичности свидетельствуют об особенностях строения молекулы. Кроме того, спектры ЯМР позволяют определить количество атомов данного типа в соединении и количество и характер атомов, окружающих рассматриваемый.
Классификация органических соединений
Существование гомологии и гомологических рядов существенно облегчает классификацию и изучение органических соединений. Гомологическим рядом называется ряд веществ, отличающихся друг от друга на любое число групп СН2, имеющих сходное строение и свойства.
В зависимости от строения углеродной цепи органические соединения делятся на соединения с открытой цепью (это алифатический, или жирный ряд) и циклические соединения. Циклические соединения могут быть карбоциклическими (циклы составлены только из атомов углерода) и гетероциклическими (циклы содержат кроме углерода и другие атомы). В свою очередь, карбоциклические соединения включают алициклический и ароматический ряды.
Наиболее простыми соединениями алифатического, алициклического и ароматических рядов являются углеводороды. Заменяя атомы водорода на другие атомы или группы ( так называемые функциональные группы), мы получаем другие классы органических соединений, свойства которых, в основном, определяет природа функциональных групп. В зависимости от наличия в соединении функциональной группы определенной природы можно выделить следующие главнейшие классы органических соединений:
углеводороды;
галогенпроизводные;
гидроксисоединения;
простые эфиры;
карбонильные соединения (альдегиды и кетоны);
карбоновые кислоты;
амины;
нитросоединения;
сульфокислоты;
элементорганические соединения.
Существует еще много классов соединений с другими функциональными группами. Кроме того, вещества могут быть насыщенными и ненасыщенными, содержать одну какую-либо функциональную группу или одновременно несколько одинаковых или различных.
Основные сырьевые источники органических соединений
Природный газ
В природном газе находятся низшие газообразные алканы – метан, этан, пропан. Содержание метана в смеси обычно доходит до 98 %. Перерабатывается преимущественно пиролизом (высокая температура) на ацетилен, водород, газовую сажу.
Нефть
Нефть – наиболее важный источник углеводородного сырья. Она представляет собой темно-коричневую вязкую маслянистую жидкость с удельной массой около 0,8 г·см-3. Среднее содержание в нефти углерода составляет 82 %…88 %, водорода – 12 %…14 %. Таким образом, нефть на 94 %…99 % состоит из смеси углеводородов. Кроме того, в небольших количествах в неё входят кислород-, серу- и азотсодержащие соединения. Ненасыщенных соединений алифатического и алициклического рядов нет. Углеводороды представлены соединениями трех классов: алканы (метановые углеводороды), цикланы (нафтены) и ароматические углеводороды. Метановые углеводороды содержатся в нефти в основном в виде н-алканов. Среди изомеров преобладают малоразветвленные. Так, метилалканов больше, чем ди- и триметилалканов. Цикланы представлены почти исключительно пяти- и шестичленными циклами, ароматические углеводороды – производными бензола. В несколько меньших количествах в нефти встречаются соединения с конденсированными кольцами циклического, ароматического и циклоароматического характера.
В нефти имеются газообразные, жидкие и твердые углеводороды. После поднятия нефти на поверхность земли от неё отделяют газообразные соединения (попутный газ), воду, механические примеси и подвергают разгонке на фракции по температурам кипения. При этом выделяют фракции авиационного и автомобильного бензинов, лигроина, керосина, солярового масла и мазута. Лигроин, керосин, соляровое масло используют в качестве горючего в дизельных двигателях. Керосин после дополнительного гидрирования служит топливом для реактивных двигателей. Мазут разгоняют под вакуумом с выделением фракций легких и тяжелых масел, которые в больших количествах используются в промышленности и транспорте. Остаток после перегонки (пёк) используется как дорожное покрытие.
В двигателях всех типов происходит сгорание углеводородов преимущественно до СО2 и Н2О. На промежуточной стадии окисления образуются гидроперекиси ROOH, которые распадаются на альдегиды, кетоны, спирты, кислоты и недолгоживущие свободные радикалы. В двигателе внутреннего сгорания при сжатии смеси паров бензина и воздуха н-алканы могут образовывать перекиси, вызывающие преждевременное воспламенение без участия запальной свечи, которая даёт искру только в момент наибольшего сжатия смеси. Явление это называется детонацией. Оно причиняет вред, т.к. двигатель быстрее изнашивается и мощность его используется неполностью. Разветвленные алканы, цикланы и особенно ароматические углеводороды в меньшей мере склонны к детонации. Детонационная стойкость бензинов оценивается октановыми числами. Октановое число изооктана (2,2,4-триметилпентана) принято за 100, н-гептана – за 0. Бензин сравнивают с искусственной смесью, которая содержит а % изооктана и (100–а)% н-гептана. При одинаковом поведении обеих проб в двигателе бензину приписывают октановое число а. Октановые числа некоторых соединений: н-пентан – 61,7; 2-метилбутан – 90,3; н-гексан – 26; н-нонан – 44; циклогексан – 77,2; бензол – 111,6; этиловый спирт – 100. Для повышения детонационной стойкости в некоторые виды бензинов добавляют небольшие количества антидетонаторов – веществ с очень высокими октановыми числами, например этиловую жидкость (смесь тетраэтилсвинца и бромистого этила), разветвленные простые эфиры, другие соединения.
С целью увеличения выхода бензина, улучшения его качества и получения сырья для нефтехимической промышленности высококипящие фракции нефти (начиная с керосина) подвергают так называемой деструктивной переработке. К процессам деструктивной переработки относятся термический и каталитический крекинги, пиролиз и риформинг. В этих процессах большие молекулы углеводородов претерпевают разрыв по С–С связи с образованием соединений с более короткой цепью углеродных атомов, соответствующих бензину. Именно эта реакция разрыва цепи характерна для термического крекинга, протекающего по радикальному механизму при температурах 400 °C…600 С и давлении 20…40 атм с образованием алканов и алкенов.
R-CH2-CH2╫CH2-CH2-CH2-CH2-CH2-CH2-CH3
R-CH2-CH3 + CH2=CH-CH2-CH2-CH2-CH2-CH3
В результате термического крекинга получается большое количество алканов и алкенов с более короткой цепочкой. Выход бензина, считая на сырую нефть, увеличивается при этом с 30 % до ~ 50 %…60 %. Однако присутствие ненасыщенных соединений (алкенов) ухудшает качество бензина.
Температура пиролиза выше: 700 °C…900 С. В этих условиях получается много ненасыщенных газообразных углеводородов с малой молекулярной массой, которые служат сырьем для нефтехимии.
Каталитический крекинг протекает при температуре 250 °C…500 С на катализаторе (например глина, обработанная кислотой и др.). Механизм цепной карбонийионный. Поскольку карбониевые ионы в отличие от радикалов более склонны к изомеризации, в результате процесса получается бензин, обогащенный изоалканами и имеющий большее октановое число. Непредельных соединений образуется меньше. Бензин более стабилен.
Качество бензина прямой гонки может быть улучшено путем каталитического риформинга. (Условия: t ~ 500С; давление 15…40 атм; катализатор: платина на Al2O3). В этом процессе алициклические углеводороды и некоторые алканы превращаются в ароматические углеводороды, которые имеют высокие октановые числа.
Каменноугольная смола
Каменноугольная смола наряду с коксом, коксовыми газами и надсмольной водой получается при коксовании каменного угля. Содержит широкий спектр ароматических и гетероциклических соединений.