

8.13. Уравнение скорости реакции нулевого порядка
Если в уравнении скорости отсутствуют члены, содержащие концентрацию реагирующих веществ, то скорость такой реакции выражается уравнением нулевого порядка
![]() . (8.26)
. (8.26)
Реакция нулевого порядка представляет особый интерес в каталитических процессах, когда катализатор, взятый в определенном количестве, может служить фактором, лимитирующим скорость реакции. Такая ситуация встречается в ферментативной кинетике.
8.14. Определение порядка реакции
Порядок реакции играет существенную роль при изучении и раскрытии механизма реакции. Он в значительной степени зависит от механизма реакции. Порядок реакции определяется опытным путем, и его нельзя предсказать заранее, даже для реакций формально похожих.
а) Для определения порядка реакции часто используют способ подстановки. Он заключается в выборе уравнения кинетики реакции (нулевого, первого, второго порядков) при подстановке в которое экспериментальных данных получается постоянное значение константы скорости реакции.
б) Существует и графический способ определения порядка реакции. Например, для реакции нулевого порядка скорость реакции не зависит от концентрации. Для реакции первого порядка прямолинейной является зависимость lnCот времени. Для реакции второго порядка линейной будет зависимость 1/Сот времени.
в) Используется также определение зависимости от концентрации константы полупревращения и т.д.
8.15. Теоретические основы химической кинетики
В основе современной химической кинетики лежат две теории: теория активных соударений и теория активного комплекса.
8.15.1. Теория активных соударений
Теория была сформулирована С. Аррениусом в 1889 году. В основе этой теории лежит представление о том, что для протекания химической реакции необходимо соударение между молекулами исходных веществ, а число соударений определяется интенсивностью теплового движения молекул, т.е. зависит от температуры. Но не каждое соударение молекул приводит к химическому превращению – к нему приводит лишь активное соударение.
Тот минимальный запас энергии, которым должны обладать молекулы исходных веществ для того, чтобы их соударение было активным, называют энергетическим барьером реакции.Наглядное представление об энергетическом барьере реакции дает графическое изображение энергетики химической реакции (рис. 8.4).
В качестве абсциссы в этих диаграммах используется так называемая координата реакции. Вообще говоря, она является сложной функцией межатомных расстояний. Но для практических целей и простых молекул можно считать, что она характеризует изменения в межатомных расстояниях, которые происходят при сближении исходных молекул, образующих активированный комплекс, и взаимном удалении продуктов реакции при распаде активированного комплекса. По оси ординат откладывается потенциальная энергия всей системы.
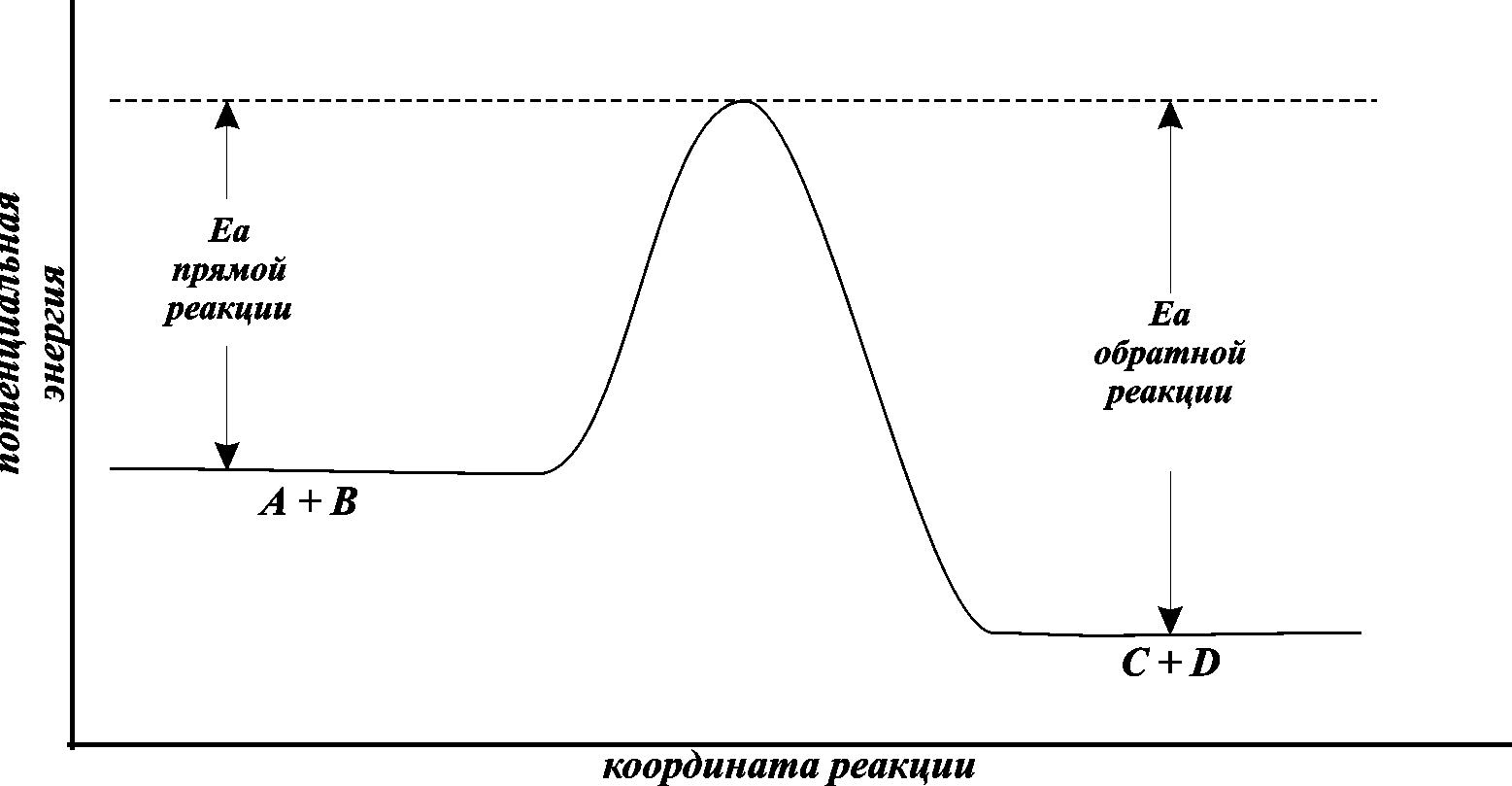
Рис. 8.4. Энергетическая диаграмма реакции А+B=C+D
То дополнительное количество энергии, которое нужно добавить к средней энергии молекул исходных веществ, чтобы соударение стало активным, называется энергией активации.
Энергия активации ощутимо влияет на значение константы скорости реакции и ее зависимости от температуры: чем больше Еа, тем меньше константа скорости и тем значительнее влияет на нее изменение температуры. Константа скорости реакции связана с энергией активации сложной зависимостью, описанной уравнением Аррениуса:
![]() ,(8.27)
,(8.27)
где А– так называемое «число соударений», которое представляет собой число соударений в одну секунду между одной молекулой вещества а и одной молекулой веществаВ, заключенных в объеме в 1см3.
Однако наблюдаемые константы скорости реакции, как правило, гораздо меньше вычисленных по уравнению. Поэтому уравнение для константы скорости реакции видоизменяют следующим образом:
![]() , (8.28)
, (8.28)
где Z– теоретическое число столкновений, аР– так называемый фактор вероятности или стерический, который учитывает все влияния, вызывающие отклонения от идеального уравнения. Необходимость ориентации может заметно тормозить даже сравнительно простые реакции. Хорошо изученным примером является реакцияН2 + J2 2НJ.
Для того, чтобы простое соударение дало две молекулы йодистого водорода, надо, чтобы ориентация молекулы была сходна с той, которая изображена на рис. 8.5 а.

Рис. 8.5. Схема, отображающая значение благоприятной ориентации для того, чтобы простое соударение могло привести к образованию продукта реакции
Энергия активации этой реакции невелика, но скорость мала. Это связано с довольно жесткими требованиями, предъявляемыми к ориентации реагирующих молекул. Тогда А = РZ, т.е.ахарактеризует число соударений с благоприятной ориентацией и называется предэкспоненциальным множителем.
Используя уравнение Аррениуса, можно определить энергию активации Еа. Для этого уравнение Аррениуса удобно применять в логарифмической форме:
![]() (8.29)
(8.29)
или
![]() . (8.30)
. (8.30)
Если построить график lgkот 1/Е, то получим прямую, отсекающую на оси ординат отрезок, равный lgА,и имеющую тангенс угла, равный ‑Еа/2,303R, т.е. tg= -Еа/2,303R, откудаЕа= -2,303Rtg.

Рис. 8.6. График Аррениуса
Из уравнения видно, что константа скорости реакции kявляется произведением двух сомножителей. Предэкспоненциальный множитель А практически не зависит от температуры, так как последняя не влияет на взаимную ориентацию молекул. Экспоненциальный множительe-Ea/RT, который характеризует долю активных соударений от общего числа двойных соударений, сильно зависит от температуры.
Теория активных соударений внесла в химическую кинетику новые представления об активных соударениях и об энергии активации, но эта теория не рассматривала механизм самого соударения, что является ее недостатком.