

Biomedical EPR Part-B Methodology Instrumentation and Dynamics - Sandra R. Eaton
.pdf272 |
CANDICE S. KLUG AND JIMMY B. FEIX |
2.1Secondary structure determination
The ability to determine and resolve local secondary structure within a protein is instrumental in the study of not only its structure, but also its functional dynamics. The SDSL approach has been used successfully to identify secondary structures in soluble proteins for a number of years. The workhorse of this and many of the other SDSL techniques has been T4 lysozyme (T4L), a relatively small, predominantly  protein that is readily expressed and purified.
protein that is readily expressed and purified.
Secondary structure determination is carried out through the use of paramagnetic probes such as oxygen, nickel complexes (e.g., nickel (II) acetylacetonate, NiAA, and nickel (II) ethylenediaminediacetate, NiEDDA), and chromium oxalate (CROX). The spin label side chain reports on its accessibility to these reagents through changes in its saturation behavior under increasing amounts of microwave power. A plot of microwave power vs. signal amplitude is fit to an equation that yields the parameter  the microwave power at which the height of the center nitroxide line is half what it would be in the absence of saturation (Altenbach et al., 1994). Accessibility of a spin label to a paramagnetic reagent decreases the effective spin-lattice relaxation time,
the microwave power at which the height of the center nitroxide line is half what it would be in the absence of saturation (Altenbach et al., 1994). Accessibility of a spin label to a paramagnetic reagent decreases the effective spin-lattice relaxation time,  resulting in an increase in the
resulting in an increase in the  value. Thus, the more accessible a spin label is to a given paramagnetic reagent, the higher the
value. Thus, the more accessible a spin label is to a given paramagnetic reagent, the higher the 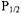 value since the power required for saturation is increased due to collisions of the spin label with the paramagnetic relaxation
value since the power required for saturation is increased due to collisions of the spin label with the paramagnetic relaxation
reagent. Assuming that any change in the spin-spin relaxation time, |
upon |
|||
addition of the relaxation agent is negligible relative to the change in |
(a |
|||
good assumption |
since |
for spin labels in biological systems at ambient |
||
temperatures is typically about an order of magnitude shorter than |
the |
|||
change in |
relative to a standard measured under |
for example |
||
|
|
is directly proportional to the |
bimolecular |
|
collision rate of the spin label with the paramagnetic probe (Altenbach et al., 1989a). To enable comparison between sites with different linewidths (i.e.,
different |
and between different laboratories, |
values are normalized |
to the peak-peak width of the center line |
and to the linewidth and |
|
saturation properties of a DPPH (diphenylpicrylhydrazyl) standard (Farahbakhsh et al., 1992), generating the accessibility parameter,  e.g.
e.g. 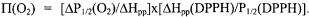 This method is termed continuous wave (CW) power saturation, and is by far the most commonly used method to determine spin label accessibility. However, it should be noted that another method involving the use of saturation recovery (SR) EPR has also been utilized that directly measures the spin lattice relaxation time of the nitroxide (e.g. (Altenbach et al., 1989b). The change in
This method is termed continuous wave (CW) power saturation, and is by far the most commonly used method to determine spin label accessibility. However, it should be noted that another method involving the use of saturation recovery (SR) EPR has also been utilized that directly measures the spin lattice relaxation time of the nitroxide (e.g. (Altenbach et al., 1989b). The change in  in the presence of a given relaxation agent provides a direct measure of
in the presence of a given relaxation agent provides a direct measure of
SDSL: A SURVEY OF BIOLOGICAL APPLICATIONS |
273 |
spin label accessibility, circumventing the need to adjust for linewidth or laboratory conditions.
Based solely on accessibility data for a given site, the general environment in which it is located can be established. For example, high accessibility to oxygen indicates a membrane-exposed site and low accessibility could indicate either solvent exposure or burial within the protein core. High accessibility to polar nickel compounds such as NiEDDA or NiAA indicates a solvent exposed site, whereas low or no accessibility would indicate a membrane exposed or a buried site. CROX is a charged polar reagent that, unlike nickel compounds, does not partition into the membrane and can be a good indicator of true solvent exposure. A combination of 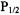 values collected for two or three of these reagents will indicate the specific environment and location of a labeled site. For instance, high
values collected for two or three of these reagents will indicate the specific environment and location of a labeled site. For instance, high  very low
very low 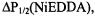 and no accessibility to CROX indicates a membrane-exposed site; low
and no accessibility to CROX indicates a membrane-exposed site; low 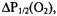 very high
very high 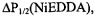 and high
and high  would indicate a solvent-exposed site; and low
would indicate a solvent-exposed site; and low  values for all reagents generally indicates a site buried within the protein structure.
values for all reagents generally indicates a site buried within the protein structure.
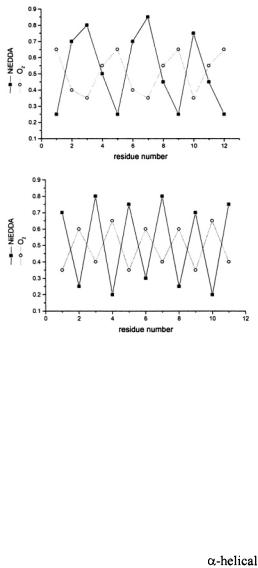
Secondary structure can then be identified by scanning through a region of the protein with the nitroxide spin label and plotting the accessibility of each site to either oxygen or nickel compounds against residue number. A periodicity in the data of ~3.6 indicates  structure due to the fact that there are 3.6 residues per turn in an
structure due to the fact that there are 3.6 residues per turn in an  Likewise, a periodicity of 2 is observed for
Likewise, a periodicity of 2 is observed for 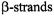 as neighboring residues are positioned on opposite faces of the strand. Oxygen and NiEDDA accessibilities for membraneembedded secondary structures are out-of-phase (Figure 2), whereas the periodicities of oxygen and NiEDDA accessibility in water-soluble protein and aqueous regions of membrane proteins exhibit in-phase periodicities. Loop regions are identified by the typical lack of any periodicity in a dataset.
as neighboring residues are positioned on opposite faces of the strand. Oxygen and NiEDDA accessibilities for membraneembedded secondary structures are out-of-phase (Figure 2), whereas the periodicities of oxygen and NiEDDA accessibility in water-soluble protein and aqueous regions of membrane proteins exhibit in-phase periodicities. Loop regions are identified by the typical lack of any periodicity in a dataset.
The  of the soluble T4L (e.g. (Mchaourab et al., 1996)) and colicin E1 (Todd et al., 1989; Salwinski and Hubbell, 1999; Vogelsang et al., 2001) proteins have been studied extensively, and the transmembrane helical bundles of bacteriorhodopsin (Altenbach et al., 1990; Altenbach et al., 1989a; Altenbach et al., 1994) and rhodopsin (Altenbach et al., 1996; Farahbakhsh et al., 1993; Farrens et al., 1996; Resek et al., 1993) were the first membrane structures studied by SDSL techniques, even prior to the publication of their crystal structures. In fact, SDSL studies were able to resolve loop regions of rhodopsin that the crystal structure was unable to detect (Langen et al., 1999). The ability of this technique to examine local structure in solution, and not just a static structure as is determined by x-ray crystallography, is one of its unique advantages. Crystal structures are excellent starting points for further analysis, but no other technique can so
of the soluble T4L (e.g. (Mchaourab et al., 1996)) and colicin E1 (Todd et al., 1989; Salwinski and Hubbell, 1999; Vogelsang et al., 2001) proteins have been studied extensively, and the transmembrane helical bundles of bacteriorhodopsin (Altenbach et al., 1990; Altenbach et al., 1989a; Altenbach et al., 1994) and rhodopsin (Altenbach et al., 1996; Farahbakhsh et al., 1993; Farrens et al., 1996; Resek et al., 1993) were the first membrane structures studied by SDSL techniques, even prior to the publication of their crystal structures. In fact, SDSL studies were able to resolve loop regions of rhodopsin that the crystal structure was unable to detect (Langen et al., 1999). The ability of this technique to examine local structure in solution, and not just a static structure as is determined by x-ray crystallography, is one of its unique advantages. Crystal structures are excellent starting points for further analysis, but no other technique can so
274 |
CANDICE S. KLUG AND JIMMY B. FEIX |
unobtrusively “watch” a protein in solution at such a detailed level. The information on molecular dynamics provided by SDSL is an excellent complement to the high-resolution structural detail provided by crystallographic studies.
Figure 2. Accessibility periodicity. Examples of out-of phase periodicity in the accessibility parameters, for
for  and NiEDDA observed for a membrane-associated
and NiEDDA observed for a membrane-associated  (top) and
(top) and  (bottom).
(bottom).
Another structure solved by SDSL methods prior to the release of a crystal structure is the Streptomyces lividans potassium channel KcsA. The organization of the transmembrane helices was studied by nitroxide scanning and accessibility measurements in the two transmembrane segments that make up each of the four monomers of KcsA (Perozo et al., 1998). It was clearly established that the first transmembrane helix (TM1) was located on the outside of the channel with TM2 lining the interior of the channel at the four-fold axis of symmetry. The publication of the crystal structure of KcsA
generally confirmed the SDSL data. |
|
Two helices have been nitroxide scanned in the |
annexin XII, a |
soluble protein that binds to lipid bilayers in the presence of calcium (Isas et al., 2002). The SDSL study confirmed the position and orientation of the

SDSL: A SURVEY OF BIOLOGICAL APPLICATIONS |
275 |
helices D and E and the connecting hairpin loop in annexin XII, which is believed to be involved in a calcium-dependent interaction with lipid bilayers. This study also demonstrated a close correlation between accessibility at a given site and the fraction of accessible surface area for the corresponding residue obtained from the crystal structure. Accessibility studies of membrane-bound annexin at pH 4.0 suggested that the helix-loop- helix motif inserted into the bilayer to form a single, membrane-spanning 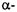
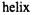 (Langen et al., 1998a).
(Langen et al., 1998a).
Nitroxide scanning accessibility measurements on residues 129–145 of the inhibitory component of the troponin complex TnI, which plays an essential role in the regulation of striated muscle contraction, indicated that
TnI was unstructured in a binary complex with the |
component |
|
TnC but formed |
secondary structure at residues 129–137 upon |
|
formation of the ternary TnI-TnC-TnT complex that interacts with tropomyosin (Brown et al., 2002). Helical secondary structure was confirmed by distance measurements between pairs of nitroxides, and changes in spin label motion upon ternary complex formation identified a tertiary contact surface. These studies conflicted with two earlier molecular models of TnI, but were in good agreement with a preliminary, unpublished crystal structure.
|
Although the |
motif was studied first and most extensively, work |
|
on |
is following closely. Early studies of soluble |
were |
|
carried out on the soluble proteins cellular retinol binding protein (CRBP) (Hubbell et al., 1998) and 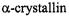 (Berengian et al., 1997; Koteiche et al., 1998). These studies established the expected periodicity of 2.0 for
(Berengian et al., 1997; Koteiche et al., 1998). These studies established the expected periodicity of 2.0 for 
accessibility (Hubbell et al., 1996). |
|
Integral membrane proteins dominated by |
structure have also |
been studied by SDSL. These include the outer membrane protein receptors FepA and BtuB. Investigations of the putative  structure of the outer membrane protein FepA were the first to identify a transmembrane
structure of the outer membrane protein FepA were the first to identify a transmembrane  by SDSL methods (Klug et al., 1997). One entire strand was structurally characterized and determined to traverse the membrane at a slight angle, consistent with
by SDSL methods (Klug et al., 1997). One entire strand was structurally characterized and determined to traverse the membrane at a slight angle, consistent with 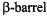 structure. The publication of the crystal structure of FepA followed soon after, confirming the SDSL results and supporting the
structure. The publication of the crystal structure of FepA followed soon after, confirming the SDSL results and supporting the
findings that the transmembrane |
had continued structure several |
|
residues beyond the surface of the membrane. In addition, the vitamin |
||
transporter, BtuB, was confirmed to have |
|
using SDSL methods |
(Fanucci et al., 2002). In this study, two consecutive |
strands were |
|
characterized and it was found that even numbered sites faced the lipid bilayer and odd numbered sites faced the channel interior. Since the crystal structure of BtuB had not yet been published, a model of the protein was constructed based on the SDSL data collected and compared to the crystal

276 |
CANDICE S. KLUG AND JIMMY B. FEIX |
data of FepA and FhuA, another outer membrane transporter. Additional studies on ligand-induced conformational changes in FepA and BtuB are discussed below.
2.2Protein-Membrane Interactions
One advantage to the study of membrane proteins is that secondary structure information can be combined with depth measurements (described below) to give information on the position and orientation of the  or
or 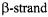 relative to the bilayer normal. Oxygen is hydrophobic and therefore has an increasing concentration gradient into the bilayer, with the maximum at the center, whereas nickel complexes are polar and have a decreasing gradient into the bilayer, with a very low concentration in the center of the bilayer and a high concentration on the surface. This reciprocal concentration gradient of commonly used reagents in the membrane allows for the estimation of depth of a spin label within the lipid bilayer.
relative to the bilayer normal. Oxygen is hydrophobic and therefore has an increasing concentration gradient into the bilayer, with the maximum at the center, whereas nickel complexes are polar and have a decreasing gradient into the bilayer, with a very low concentration in the center of the bilayer and a high concentration on the surface. This reciprocal concentration gradient of commonly used reagents in the membrane allows for the estimation of depth of a spin label within the lipid bilayer.
Spin labeled lipids with the labels at given depths are used to calibrate each bilayer system. The accessibilities of the labels to oxygen and
NiEDDA are recorded and converted to a value, |
which is defined by the |
|
natural log of the ratio of the oxygen and nickel accessibilities: |
||
(Altenbach et al., |
1994). |
is then plotted |
against the known depths of the nitroxides within the bilayer to give a calibration equation for further depth experiments: 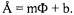
In addition to the relatively early, seminal studies on the 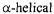 transmembrane proteins rhodopsin and bacteriorhodopsin mentioned above, SDSL has contributed to the understanding of membrane insertion by channel-forming proteins such as colicin E1 and diphtheria toxin.
transmembrane proteins rhodopsin and bacteriorhodopsin mentioned above, SDSL has contributed to the understanding of membrane insertion by channel-forming proteins such as colicin E1 and diphtheria toxin.
The structure of two consecutive helices from the channel forming toxin colicin E1 was studied by SDSL. Twenty three residues were scanned with a spin label and it was found that they formed two separate helices connected by a hairpin loop, a configuration that closely matched that found in the crystal structure of the channel-forming domain (Salwinski and Hubbell, 1999). However, upon binding to negatively charged membranes at low pH (pH4.0), these two colicin E1 helices form one long helix that appears to insert into the lipid bilayer. One face of the helix was found to face a polar environment and the other a hydrophobic environment. In order to distinguish whether the helix was laying across the top of the bilayer or inserted into it, depth measurements were carried out that identified this helix as being transmembrane. Further studies on the structure of neighboring helices reveal that they also extend into one longer helix, but that upon binding to bilayers at low pH the extended helices localize to the membrane/water interface (Vogelsang et al., 2001).
SDSL: A SURVEY OF BIOLOGICAL APPLICATIONS |
277 |
Twenty-one consecutive residues in a putative transmembrane helix (TH9) of diphtheria toxin were analyzed in the presence of phospholipid vesicles at pH 4.6, corresponding to the endosomal pH at which translocation of the toxin occurs (Oh et al., 1996). The pattern of accessibility to 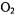 and NiEDDA indicated that this segment was
and NiEDDA indicated that this segment was 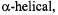 with one side of the helix facing the lipid and the other exposed to the aqueous phase. Depth measurements using SDSL on those sites facing the bilayer indicated a transmembrane orientation of the helix.
with one side of the helix facing the lipid and the other exposed to the aqueous phase. Depth measurements using SDSL on those sites facing the bilayer indicated a transmembrane orientation of the helix.
Accessibility measurements have also played an important role in determining secondary structure and penetration depth for a number of smaller, membrane-active peptides. Although such peptides are amenable to structure determination by NMR and secondary structure analysis by CD spectroscopies, these methods do not provide the detailed information on interaction with the lipid bilayer that is available from SDSL studies. Important examples include alamethicin (Archer et al., 1991; BarrangerMathys and Cafiso, 1996; Lewis and Cafiso, 1999) and the 25-residue MARCKS (myristoylated alanine-rich protein kinase C substrate) peptide. Depth measurements for twelve single-cysteine analogs indicated that MARCKS is oriented along the bilayer surface with its N-terminus extending into the aqueous phase (Qin and Cafiso, 1996). Additional studies on the interaction of MARCKS with phosphatidylinositol are discussed below. A peptide derived from the protein kinase C/calmodulin binding domain of neuromodulin also bound to negatively-charged bilayers in an extended conformation along the membrane surface with its N- and C- termini extended into the aqueous phase, although the central region of this peptide penetrated more deeply into the hydrophobic phase than MARCKS (Wertz et al., 1996). In contrast, an N-terminal myristoylated peptide derived from Src bound to membranes with its N-terminus close to the bilayer interface and the C-terminal half extended into the aqueous phase (Victor and Cafiso, 1998).
A membrane-binding presequence of yeast cytochrome c oxidase was bound to membranes in an extended conformation, with the nitroxide side chains immersed approximately 13Å below the lipid headgroups (Yu et al., 1994). Accessibility measurements on the N-terminal fusion peptide of influenza virus hemagglutinin indicated that it inserted into the bilayer as an  tilted ~ 25° relative to the membrane surface (Macosko et al., 1997).
tilted ~ 25° relative to the membrane surface (Macosko et al., 1997).
An extensive study has recently examined depth of penetration and orientation of the C2 domain of cytosolic phospholipase A2 when bound to membranes in the presence of 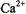 (Frazier et al., 2002). Values of
(Frazier et al., 2002). Values of  were combined with constraints from the solution NMR structure to generate a model for the protein at the membrane interface. It was suggested that the dependence of
were combined with constraints from the solution NMR structure to generate a model for the protein at the membrane interface. It was suggested that the dependence of  on bilayer depth is best represented by a hyperbolic tangent
on bilayer depth is best represented by a hyperbolic tangent
278 CANDICE S. KLUG AND JIMMY B. FEIX
function rather than the linear relationship described above, especially near the bilayer-aqueous interface. This is intuitively consistent with the fact that concentrations of relaxation agents must reach some limiting value in the bulk solution and in an infinitely deep membrane.
Docking of the soluble bee venom phospholipase A2 to small unilamellar vesicles of non-hydrolyzable, anionic phospholipids was studied by measuring the accessibility of several sites to CROX in the presence and absence of membranes (Lin et al., 1998). The concentration of this negatively-charged relaxation agent is expected to decrease near the bilayer surface due to electrostatic repulsion, providing relative distances to the various spin-labeled sites. These relative distances were in turn used to
orient the protein, based on its known crystal structure. |
|
In addition to measurement of relative accessibilities to |
and NiEDDA, |
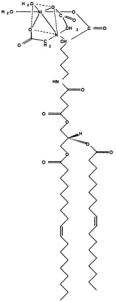
a novel methodology has recently been published for determining the location of residues facing the membrane-aqueous interface (Gross and Hubbell, 2002). The ability to identify residues not only within the membrane bilayer, but also those near the membrane-aqueous interface is invaluable to identification of global protein structure and folding because it restricts the number of possible orientations of the entire protein by confining specific residues to specific surroundings. This technique involves introducing a Ni-chelating lipid (DOGS-NTA; Figure 3) into the membrane and measuring the accessibility of the protein-attached spin label to the paramagnetic nickel ion. Since the metal chelate is attached to the lipid through a spacer, there is an approximately 14Å band near the membrane surface where collisions can occur, which provides an excellent starting point for more accurate positioning. This is especially useful in the absence of a crystal structure.
2.3Conformational changes in secondary structure
Conformational changes in protein structure are often reflected in both accessibility measurements and in motional changes and therefore cannot strictly belong to one category of SDSL techniques. Illustrated here are two examples of accessibility measurements as a tool for the study of conformational changes; more examples can be found in the sections on motion and distance measurements.
Lactose permease is an  membrane protein involved in the translocation of galactosides and has been extensively studied by SDSL (e.g. (Zhao et al., 1999; Sun et al., 1999; Wang et al., 1998)). Many of the helices have been characterized by nitroxide scanning, and conformational changes have been identified through changes in solvent accessibility (Zhao et al., 1999). Upon nitroxide scanning of helices IV and V, it was found that
membrane protein involved in the translocation of galactosides and has been extensively studied by SDSL (e.g. (Zhao et al., 1999; Sun et al., 1999; Wang et al., 1998)). Many of the helices have been characterized by nitroxide scanning, and conformational changes have been identified through changes in solvent accessibility (Zhao et al., 1999). Upon nitroxide scanning of helices IV and V, it was found that
SDSL: A SURVEY OF BIOLOGICAL APPLICATIONS |
279 |
binding of the ligand, 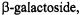 had no apparent effect on the motion of the labeled side chains. However, an unexpected increase in solvent accessibility at site 137 on helix IV and a decrease in solvent accessibility on one entire face of helix V after ligand binding indicated a translational motion of the entire helix V, yet without perturbation of side chain motion.
had no apparent effect on the motion of the labeled side chains. However, an unexpected increase in solvent accessibility at site 137 on helix IV and a decrease in solvent accessibility on one entire face of helix V after ligand binding indicated a translational motion of the entire helix V, yet without perturbation of side chain motion.
Figure 3. DOGS-NTA.
The mechanosensitive channel, MscL, is a homopentamer of monomers comprised of two transmembrane helices and a cytoplasmic helix that are all thought to be involved in channel gating. It was found by SDSL investigations that a large rearrangement of the monomers occurs upon lipid composition-induced channel opening (Perozo et al., 2002a). Structures of the channel domain in the closed, intermediate, and open states were all determined by SDSL accessibility measurements and the pore size was found to increase from essentially closed to greater than 25Å wide in the open state. Data indicating increases in solvent accessibility and the dynamics of the side chains in both transmembrane helices and the absence of spin-spin interactions between the monomers were combined to map out the nature of the channel rearrangement upon gating.

280 |
CANDICE S. KLUG AND JIMMY B. FEIX |
3.MOTION
The ability to introduce a spin label side chain into a protein at very specifically chosen locations allows the study of the local motion of a particular site within a protein structure. EPR spectra are sensitive to the overall tumbling of the protein, the local motion of the spin label side chain, and the motion of the protein backbone. In order to observe only the motion of interest, it is possible to slow the tumbling of small proteins with viscous solvents (e.g., 30% sucrose, see below), thereby leaving the spectrum sensitive to only side chain and backbone dynamics. In addition, rotation of the bonds within the spin label itself can be dampened to leave the spectrum sensitive to only the backbone motion of the protein. The spectral information gained based on motion alone is another important feature of SDSL and a number of the protein systems that have benefited from this technique are discussed below. Since packing of the nitroxide side chain within the protein structure affects its motion, a good deal of information can be gained on local tertiary and/or quaternary interactions based on motion alone. A particular strength of the spin labeling technique is its ability to report on changes on very localized dynamics. Thus, conformational changes due to substrate binding, membrane binding, secondary, tertiary and protein-protein interactions, denaturation, and other perturbations are all evident in the EPR spectra and can give key information on structural changes at specific sites.
Rotational motion influences the spin label EPR spectrum primarily through modulation of hyperfine and g-tensor anisotropies (Stone et al., 1965; Nordio, 1976), leading to changes in line amplitudes, widths, and positions. The term motion encompasses both the rate and amplitude of changes in the orientation of the spin label in the external magnetic field and rigorous motional analysis requires spectral simulation. Nonetheless, it is often possible to obtain biologically relevant estimates of changes in motion and patterns of motional freedom from readily accessible spectral
parameters. |
|
The inverse width of the center line, |
has proven to be a |
convenient, semi empirical parameter for the estimation of relative rotational mobility (Hubbell et al., 2000; Hubbell et al., 1998; Hubbell et al., 1996). It is particularly valuable for determination of secondary structure when
scanning a continuous region of a protein. For example, |
values for |
||
sites in a known |
of phage T4L |
show a periodicity that closely |
|
matches the 3.6 residue/turn of an ideal |
(Mchaourab et |
al., 1996). |
|
Similar observations have been made for a large number of systems, including colicin E1 (Salwinski and Hubbell, 1999), lactose permease (Voss et al., 1997), and tear lipocalins (Glasgow et al., 1999). Such patterns in
SDSL: A SURVEY OF BIOLOGICAL APPLICATIONS |
281 |
rotational mobility generally match quite closely with those obtained from accessibility measurements, but tend not to apply to motionally restricted sites engaged in tertiary contacts.
3.1Local side chain motion
It has been shown in phage T4L, one of the proteins most thoroughly studied by SDSL, through the study of a large number of mutants by X-ray crystallography (e.g., (Matthews, 1995)) that this protein tolerates single-site mutations without global changes in tertiary structure. In one study (Mchaourab et al., 1996), 30 single cysteine mutants were examined and classified based on the crystal structure of T4L as 1) solvent exposed helical sites not in the N- or C-cap regions, 2) solvent-exposed sites near the N- or C-termini of  3) sites with at least some degree of tertiary interaction, 4) sites in solvent-exposed loops, and 5) sites buried in the hydrophobic core with no solvent accessibility. Since T4L is a relatively small protein (~17 kDa), rotational tumbling of the entire molecule was suppressed by recording spectra in 30% sucrose (increasing viscosity by a factor of ~ 3) to allow examination of side chain and backbone dynamics.
3) sites with at least some degree of tertiary interaction, 4) sites in solvent-exposed loops, and 5) sites buried in the hydrophobic core with no solvent accessibility. Since T4L is a relatively small protein (~17 kDa), rotational tumbling of the entire molecule was suppressed by recording spectra in 30% sucrose (increasing viscosity by a factor of ~ 3) to allow examination of side chain and backbone dynamics.
The first crystal structures of a spin labeled protein were recently published that revealed the orientation of the spin label MTSL attached to various sites on T4L (Langen et al., 2000). Three helix surface sites and one tertiary contact site were presented. This was an important step in linking the side chain dynamics revealed through spectral analysis with a picture of the local structural orientation(s) of the spin label attached to the protein. Most importantly, the preferred conformations of the spin label side chain were correlated with their spectral fingerprints.
In all cases, the electron density of the side chains were clearly resolved, indicating that distinct orientations of the side chain are preferred in these cases. For example, at site 119, an interior helix site with a two-component spectrum, two distinct conformations of the side chain were revealed, accounting for the two spin populations observed in its EPR spectrum. Other sites gave information on molecular and tertiary contact interactions of the side chain that may give rise to its immobilization. Clearly, as additional crystal structures of spin-labeled proteins become available, the relationship between the structure of the protein and side chain mobility will be better understood, allowing correlation of the possible structural interactions with spectral mobility. This will especially benefit future spectral simulations of side chain motion and dynamics in addition to paving the way for the development of novel spin label side chains specifically designed for motional investigations.