

3.8. Термодинамическая энтропия
Понятие термодинамической энтропии, впервые введенное в 1865 году Клаузиусом, имеет ключевое значение для понимания основных положений термодинамики.
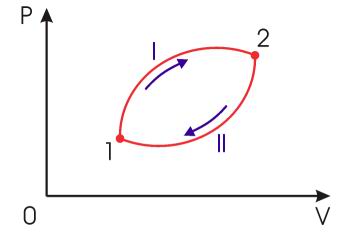
Рассмотрим обратимый круговой термодинамический процесс, представленный на рис. 3.12. Для этого процесса может быть записано равенство Клаузиуса (3.49) в виде
|
(3.50) |
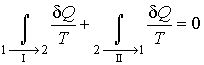 ,
,
где
первый интеграл берется по траектории ![]() ,
а второй - соответственно по траектории
,
а второй - соответственно по траектории ![]() .
.
|
Рис. 3.12. Обратимый круговой термодинамический процесс |
Изменение
направления протекания процесса ![]() на
противоположное
на
противоположное ![]() ,
что можно выполнить вследствие обратимости
процесса
,
приводит к замене знака перед вторым
интегралом формулы (3.50).
Выполнение этой замены и перенос второго
интеграла в выражении (3.50) в
правую часть дают
,
что можно выполнить вследствие обратимости
процесса
,
приводит к замене знака перед вторым
интегралом формулы (3.50).
Выполнение этой замены и перенос второго
интеграла в выражении (3.50) в
правую часть дают
|
(3.51) |
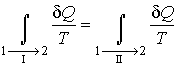 .
.
Из
полученного выражения следует, что для
обратимых процессов интеграл 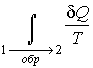 не
зависит от конкретного вида траектории,
по которой происходит процесс, а
определяется только начальным и конечным
равновесными состояниями термодинамической
системы.
не
зависит от конкретного вида траектории,
по которой происходит процесс, а
определяется только начальным и конечным
равновесными состояниями термодинамической
системы.
С аналогичной ситуацией мы уже встречались, когда в механике рассматривали определение работы консервативной силы. Независимость работы консервативной силы от формы траектории движения тела позволила ввести функцию, названную потенциальной энергией, которая зависит только от состояния механической системы и не зависит от того, как в это состояние система была переведена.
Из
этой аналогии следует, что элементарное
приведенное количество теплоты ![]() должно
представлять собой полный дифференциал
некоторой функции
должно
представлять собой полный дифференциал
некоторой функции ![]() ,
зависящей только от состояния
термодинамической системы, то есть:
,
зависящей только от состояния
термодинамической системы, то есть:
|
(3.52) |
Тогда
интеграл 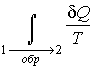 будет
равен разности значений функции
в
равновесных состояниях 1 и 2:
будет
равен разности значений функции
в
равновесных состояниях 1 и 2:
|
(3.53) |
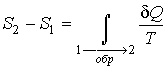 .
.Итак, величина является функцией, зависящей только от равновесного состояния термодинамической системы. Она не зависит от конкретного вида термодинамического процесса, приведшего систему в указанное состояние. Эта функция была названа Клаузиусомтермодинамической энтропией. Выражения (3.52) и (3.53) дают математическую формулировку сформулированного выше определения термодинамической энтропии.
Из выражения (3.53) следует, что термодинамическая энтропия, так же как и потенциальная энергия, определяется с точностью до произвольной постоянной. Это связано с тем, что формула (3.53) не позволяет определить абсолютное значение термодинамической энтропии, а дает только разность энтропий для двух равновесных состояний, как суммарную приведенную теплоту в обратимом термодинамическом процессе, переводящим систему из одного состояния в другое.
Термодинамическая энтропия, введенная выше, применима для описания равновесного состояния термодинамической системы. Для нахождения энтропии термодинамической системы, находящейся в квазиравновесном состоянии, при котором можно считать, что её отдельные части (подсистемы) находятся в состоянии равновесия, можно воспользоваться свойством аддитивности энтропии:
|
(3.54) |
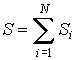 ,
,
где: ![]() -
энтропии подсистем,
-
энтропии подсистем, ![]() -
число подсистем.
-
число подсистем.
Следовательно, термодинамическая энтропия макроскопической системы, состоящей из находящихся в равновесии подсистем, равна сумме энтропий этих подсистем.
Свойство аддитивности энтропии позволяет описывать состояния макроскопической системы, не находящейся в равновесии, путем её разбиения на достаточно большое число подсистем, которые можно считать находящимися в состоянии локального равновесия. Такой подход дает возможность распространить результаты равновесной термодинамики на системы, находящиеся в неравновесном состоянии, но которые можно представить как состоящие из некоторого числа равновесных подсистем.
Задача
3.6. Жесткий теплоизолированный сосуд
объемом ![]() разделен
на две части с помощью перегородки. В
начальный момент с одной стороны
перегородки в объеме
разделен
на две части с помощью перегородки. В
начальный момент с одной стороны
перегородки в объеме ![]() находится
находится ![]() молей
идеального газа с теплоемкостью
молей
идеального газа с теплоемкостью ![]() при
температуре
при
температуре ![]() ,
а с другой стороны - в объеме
,
а с другой стороны - в объеме ![]() (
(![]() )
находится
)
находится ![]() молей
другого идеального газа с теплоемкостью
молей
другого идеального газа с теплоемкостью ![]() при
температуре
при
температуре ![]() .
Перегородку медленно удаляют, что
приводит к смешиванию газов и выравниванию
их температуры. Определить изменение
энтропии в этом процессе.
.
Перегородку медленно удаляют, что
приводит к смешиванию газов и выравниванию
их температуры. Определить изменение
энтропии в этом процессе.
Решение: Так как сосуд является жестким и теплоизолированным, то можно считать, что суммарная внутренняя энергия системы не изменяется. Тогда установившуюся температуру можно найти из соотношения:
![]() .
.
Отсюда имеем:
 .
.
Будем считать, что рассматриваемый процесс смешивания газов и выравнивания их температур является квазиравновесным. Тогда в соответствии с формулой (3.52) и первым началом термодинамики можно записать
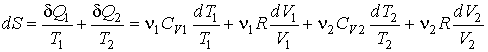 .
.
Интегрирование этого выражения в соответствии с формулой (3.53) дает:
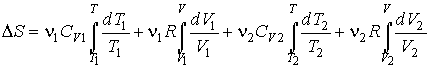
или после вычисления интегралов имеем
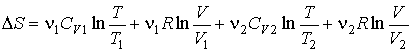 .
.
В
частном случае, если ![]() и
и ![]() имеем:
имеем:
![]() ,
,
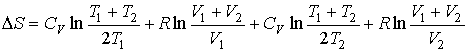 .
.
Если ![]() и
и ![]() ,
то
,
то
![]() .
.
Таким образом, при смешивании двух газов энтропия системы увеличивается как вследствие выравнивания их температуры, так и за счет их взаимного перемешивания. Оба эти процесса являются необратимыми, что и описывается возрастанием суммарной энтропии системы
. Обратимый процесс. В случае обратимого процесса
![]()
Интеграл по замкнутому контуру - это изменение энтропии во всем цикле, т.е. при обратимых циклах энтропия не меняется:
![]() 2.
Необратимый процесс.
2.
Необратимый процесс.
Исходя из определения энтропии можем записать,
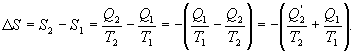 С
другой стороны для необратимого
процесса
С
другой стороны для необратимого
процесса
![]() Следовательно
Следовательно
![]() или
с учетом обратимого процесса
или
с учетом обратимого процесса
|
(9.32) |
Таким образом, энтропия изолированной системы может только возрастать (если в системе протекают необратимые процессы), либо остается постоянной (если в системе протекает обратимый процесс). Убывать энтропия изолированной системы не может.
Вопрос № 31
Статистическая интерпретация энтропии
Каждому состоянию системы приписывается термодинамическая вероятность (определяемая как число микросостояний, составляющих данное макросостояние системы), тем большая, чем более неупорядоченным или неопределенным является это состояние. Энтропия – функция состояния, описывающая степень неупорядоченности системы.
S = klnW – формула Больцмана.
Система стремится самопроизвольно перейти в состояние с максимальной термодинамической вероятностью.
Расчет абсолютной энтропии
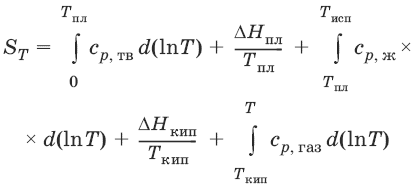
Изменение энтропии в ходе химического процесса определяется только видом и состоянием исходных веществ и продуктов реакции и не зависит от пути реакции:
ΔS = Σ(νiSi) прод – Σ(νiSi)исх
Величины абсолютной энтропии в стандартных условиях приведены в справочной литературе.
С точки зрения физики, второе начало термодинамики применимо для замкнутых систем (система считается замкнутой, если у неё отсутствует энергетический обмен с внешним миром). Открытые (т.н. "диссипативные") системы, изучением которых занимается неравновесная термодинамика, основателем которой был русский учёный И. Пригожин, второму началу термодинамики не подчиняются. Действительно, можно представить себе ситуацию, когда в систему можно внести упорядочение извне, либо система имеет предпосылки к самоорганизации. В этом случае энтропия будет уменьшаться.
Вопрос № 32
Реальные газы. Уравнение Ван-дер-вальса
РЕА́ЛЬНЫЙ ГАЗ, газ, свойства которого существенно зависят от взаимодействия молекул (см. Межмолекулярное взаимодействие).
В обычных условиях, когда средняя потенциальная энергия взаимодействия молекул много меньше их средней кинетической энергии, свойства реальных газов незначительно отличаются от свойств идеального газа и к реальным газам применимы законы, установленные для идеального газа. Отличие свойств реального газа от свойств идеального становится особенно значительным при высоких давлениях и низких температурах, когда начинают проявляться квантовые эффекты.
В модели идеального газа не учитывается собственный объем молекул и силы межмолекулярного взаимодействия. Тщательная экспериментальная проверка газовых законов (закон Бойля —Мариотта, закон Шарля, закон Гей-Люссака) современными методами показала, что эти законы достаточно точно описывают поведение реальных газов при небольших давлениях и высоких температурах. При других условиях наблюдаются значительные отступления от этих законов. Причина заключается в том, что, во-первых, при очень сильном сжатии газов объем незанятого молекулами пространства становится сравним с объемом, занимаемым самими молекулами; а во-вторых, при низких температурах становится заметным взаимодействие между молекулами. Поэтому для описания поведения газа при достаточно больших плотностях (больших давлениях) уравнения состояния идеального газа не пригодны. Наличие сил межмолекулярного взаимодействия, а именно сил отталкивания, действующих на малых расстояниях порядка размеров молекул, и сил притяжения, приводит к сложной зависимости энергии потенциального взаимодействия молекул от расстояния.
Для описания термодинамических свойств реальных газов используются различныеуравнения состояния. При малых плотностях наличие межмолекулярного взаимодействия учитывается вириальным уравнением состояния реального газа:
pV = RT[1 + B(T)/v + C(T)/v2 + ...],
где p — давление, v — мольный объем, Т — абсолютная температура, R — газовая постоянная, В(Т), С(Т) и т. д. — вириальные коэффициенты, зависящие от температуры и характеризующие парные, тройные и т. д. взаимодействия частиц в газе. Качественно верно описывает основные отличия реального газа от идеального уравнение Ван-дер-Ваальса, учитывающее существование сил притяжения между молекулами, действие которых приводит к уменьшению давления газа, и сил отталкивания, препятствующих безграничному сжатию газа.
Кроме уравнения Ван-дер-Ваальса было предложено много других эмпирических уравнений состояния реальных газов. Некоторые из них дают лучшее согласие с опытом за счет большого числа входящих в них феноменологических постоянных. Однако при качественном исследовании поведения реальных газов использование уравнения Ван-дер-Ваальса более удобно, благодаря его простоте и понятному физическому смыслу.
Одной из основных характеристик реальных газов являются размеры молекул. В реальных газах их называют газокинетическими радиусами, и их размер связан с характерными расстояниями, на которых проявляются силы межатомных и межмолекулярных взаимодействий.
В реальных газах возникают неоднородности полей давления и температуры, а также макроскопические потоки, которые приводят к переносу массы — диффузии. Для реальных газов характерна теплопроводность и вязкость. Главная особенность кинетических процессов переноса в реальных газах (в отличие от жидкостей и твердых тел) — наличие механизма столкновения молекул. Поэтому основной характеристикой этих процессов в газах является длина свободного пробега.
Внутренняя энергия реального газа зависит от объема V, то есть от расстояния между молекулами, так как потенциальная энергия молекул определяется их взаимным расположением.
Существование межмолекулярного взаимодействия в той или иной степени сказывается на всех свойствах реальных газов.
ВАН-ДЕР-ВА́АЛЬСА УРАВНЕ́НИЕ, уравнение состояния, описывающее свойства реального газа. Предложено Й. Д. Ван-дер-Ваальсом в 1873 г. Широко используется для качественного анализа поведения реальных газов и жидкостей. В модели реального газа Ван-дер-Ваальса молекулы рассматриваются как абсолютно твердые слабо притягивающиеся упругие сферы определенного диаметра.
Уравнение Ван-дер-Ваальса количественно определяет свойства реальных газов лишь в небольшом интервале температур и давлений: в области относительно высоких температур и низких давлений, так как входящие в него экспериментально определяемые константы являются функциями температуры.
Для моля газа объемом V при температуре Т и давлении р, уравнение Ван-дер-Ваальса имеет вид:
(p+a/Vm 2)(Vm - b) = RT,
где: R — газовая постоянная,
a и b — экспериментальные константы, учитывающие отклонение свойств реального газа от свойств идеального газа.
Член a/V2 имеет размерность давления и учитывает притяжение между молекулами газа за счет ван-дер-ваальсовых сил. Действие сил притяжения газа приводит к появлению дополнительного давления на газ, называемого внутренним давлением. По вычислениям Ван-дер-Ваальса, внутреннее давление обратно пропорционально квадрату молярного объема, т. е. рвн = a/Vm2, где а — постоянная Ван-дер-Ваальса, характеризующая силы межмолекулярного притяжения, Vm — молярный объем.
Константа b является поправкой на собственный объем молекул газа и учитывает отталкивание молекул на близких расстояниях. Наличие сил отталкивания, которые противодействуют проникновению в занятый молекулой объем других молекул, приводит к тому, что фактически свободный объем, в котором будут двигаться молекулы реального газа, будет не Vm, а Vm-b, где b — объем, занимаемый самими молекулами. Объем b равен учетверенному собственному объему молекул.
Константы а и b обычно определяются из экспериментальных данных, и эти величины постоянны для каждого газа. Для их определения записывают уравнения для двух известных из опыта состояний газа и решают эти уравнения относительно а и b. При больших объемах V можно пренебречь обеими поправками и уравнение Ван-дер-Ваальса переходит в уравнение состояния идеального газа (см. Клапейрона уравнение).
Несмотря на то, что уравнение Ван-дер-Ваальса является приближенным и количественно описывает свойства реальных газов лишь в области высоких температур и низких давлений, качественно оно позволяет описывать поведение газа и при высоких давлениях, конденсацию газа в жидкость. Уравнение Ван-дер-Ваальса также описывает критическое и метастабильное состояние системы жидкость-пар