

6. Замещение одних атомов галогена на другие
Среди органических галогенидов наиболее доступными являются хлориды и бромиды. Фториды и иодиды в основном получают, используя реакцию замещения одних атомов галогена на другие:
![]()
![]()

Глава 6. Процессы алкилирования
Алкилированием называются процессы замещения атома водорода или металла в молекуле субстрата на алкил. Обычно различают С-, N- и О-алкилирование, которые несколько отличаются по условиям проведения процесса. Если в молекулу вводится арил, реакция называется арилированием.
В качестве алкилирующих агентов используют главным образом, галогенпроизводные, непредельные соединения, спирты, простые эфиры и эфиры серной и сульфокислот.
Процессы алкилирования используют для построения углеродного скелета молекулы, а также временной защиты функциональных групп (чаще всего гидроксильной или аминогрупп при получении пептидов, антибиотиков или модификации сахаров). В связи с этим они имеют большое значение в химическом синтезе лекарственных веществ и витаминов.
Реакции С-, N- и О-алкилирования аренов и алифатических соединений сильно отличаются. Единого механизма, охватывающего все случаи этого процесса, нет.
1. Алкилирование аренов по Фриделю-Крафтсу
С-Алкилирование аренов можно проводить по Вюрцу-Фиттигу, Вюрцу-Гриньяру и т.д., но в промышленности чаще всего используется реакция Фриделя-Крафтса, которую можно представить в виде следующей схемы:
![]()
Механизм реакции алкилирования аренов по Фриделю-Крафтсу представляет собой обратимое электрофильное замещение (SE):
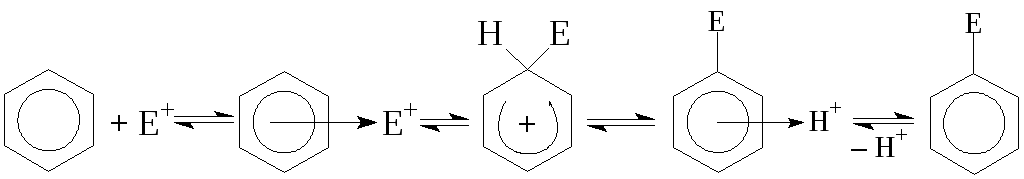
Реакционная способность субстрата определяется устойчивостью -комплекса. Как и в других реакциях SE (сульфирования, нитрования, галогенирования, нитрозирования и т. п.) электронодонорные заместители в ядре, стабилизирующие -комплекс, ускоряют реакцию, а электроноакцепторные — дестабилизируют его и затрудняют реакцию.
В связи с тем, что реакция обратима, направление реакции в мягких условиях определяется устойчивостью -комплекса (кинетический контроль) и выполняются правила ориентации. При высокой температуре, большом количестве катализатора и продолжительном времени ведения процесса направление реакции определяется устойчивостью конечных продуктов (термодинамический контроль), что часто приводит к получению метазамещенных продуктов. Например, при метилировании толуола метилхлоридом при 0 °С образуется 27 % м-ксилола, при 55 °C— 87 %, а при 106 °С — 98 %.

В качестве катализаторов в процессах алкилирования в основном применяют протонные и апротонные кислоты, оксиды, цеолиты и катиониты.
Протонные кислоты используются главным образом при алкилировании ароматических соединений спиртами и алкенами. Их активность как катализаторов падает в ряду HF > H2SO4 > H3PO4.
Апротонные кислоты (кислоты Льюиса) используются чаще всего при алкилировании алкилгалогенидами и алкенами. По активности их можно расположить в следующий ряд: AlBr3 > AlCl3 > FeCl3 >… > ZnCl2.
Оксиды металлов и бора (B2O3, Al2O3, Cr2O3 и др.), модифицированные F3,BF3.
Цеолиты имеют общую формулу M2/nО.Al2O3.xSiO2.yH2O, где М — металл, а n — его валентность. Каталитические свойства цеолитов можно менять, изменяя объем пор методом ионного обмена; алюмосиликатный состав и др.методами.
Катиониты представляют собой полимеры (чаще всего полистирольные), содержащие –SO3H, –COOH, –PO(OH)2 и другие группы.
Активность катализатора зависит также от строения субстрата, природы алкилирующего агента и условий реакции (температура, давление и т.д.). Например, трифторид бора является активным катализатором при алкилировании спиртами, алкенами, фторпроизводными, но в реакциях с другими алкилгалогенидами его активность мала.
Катализ может быть твердофазным и жидкофазным. Использование твердых гетерофазных катализаторов (оксидов, цеолитов, катионитов) предпочтительно, так как при этом упрощается технология процесса (отделение и регенерация катализатора); уменьшаются затраты на подготовку сырья, промывку реакционной массы и нейтрализацию кислых сточных вод; уменьшается коррозия оборудования; упрощается организация непрерывных процессов и т.д. В случае апротонных кислот, выбор того или иного вида катализа зависит от растворителя и свойств кислоты Льюиса. При наличии двух жидких фаз (кислотно-солевой и органической) реакция, в основном, проходит в кислотно-солевом слое.
При алкилировании с помощью алкилгалогенидов и алкенов обычно достаточно небольшого количества катализатора, а при использовании спиртов необходимо, по меньшей мере, эквимолярное количество кислоты Льюиса, так как вода, образующаяся в результате реакции, дезактивирует катализатор.
Основными недостатками реакции Фриделя-Крафтса являются полиалкилирование, изомеризация радикала, изомеризация и диспропорционирование продукта.
Полиалкилирование объясняется большей реакционной способностью продуктов алкилирования, чем исходного субстрата:

Для увеличения выхода моноалкиларена реакцию ведут в избытке субстрата при возможно более низкой температуре.
Изомеризация радикала вызывается превращением карбокатиона, образующегося в ходе взаимодействия галогенида с катализатором, в более стабильную частицу. Например, алкилирование бензола пропилбромидом в той или иной степени приводит к синтезу изопропилбензола, т.к. пропилкатион изомеризуется в более стабильный вторичный катион:

Чтобы избежать изомеризации радикала, следует тщательно подбирать условия проведения реакции (кислоту Льюиса, растворитель, время выдержки и температуру).
Изомеризация продукта реакции связана с обратимостью процесса и смещением равновесия в сторону более устойчивых веществ. Например, при нагревании п-ксилола с хлористым водородом и AlCl3, протон, выступая в качестве электрофила, атакует п-положение субстрата и вытесняет карбокатион. Продуктами взаимодействия нового электрофила (СН3+) с толуолом являются о-, м- и п-ксилолы, среди которых о- и п-изомеры сзаимодействуют с протоном и вновь превращаются в толуол, а м-ксилол накапливается в реакционной массе. Таким образом большая часть углеводорода превращается в термодинамически более устойчивый м-ксилол, в котором метильные группы в наименьшей степени вытесняются протоном:

В ряде случаев использование в процессах алкилирования веществ, связывающих образующийся галогеноводород, предотвращает обратимость реакции, а, следовательно, и изомеризацию продукта.
Диспропорционирование продуктов алкилирования также связано с обратимостью реакции, поэтому одновременно с изомеризацией продукта может проходить и перемещение алкильных групп в более замещенный арен:

В приведенном выше примере метилкатион в первую очередь реагирует с более активным чем толуол ареном (например, п- или м-ксилолом), поэтому в реакционной массе накапливаются моно- и триалкилбензолы.
Изомеризация и диспропорционирование конечного продукта в кислой среде может служить доказательством обратимости процесса алкилирования.