Группа заболеваний –Мукополисахаридозы
Мукополисахаридо́зы (МПС)— группа метаболических заболеваний соединительной ткани, связанных с нарушением обмена кислых гликозаминогликанов (мукополисахаридов), связанных недостаточностью лизосомных ферментов обмена гликозаминогликанов. Заболевания вызваны наследственными аномалиями обмена, проявляются в виде лизосомной болезни накопления: различных дефектов костной, хрящевой, соединительной тканей.
В зависимости от недостаточности одного из ферментов лизосом, накапливаются мукополисахариды одного из трёх классов: гепаран-, дерматан- или кератансульфаты.
Подавляющее большинство мукополисахаридозов (практически все) наследуются по аутосомно-рецессивному типу. Исключение составляет болезнь Хантера (мукополисахаридоз II типа), которая наследуется по X-сцепленному рецессивному механизму.
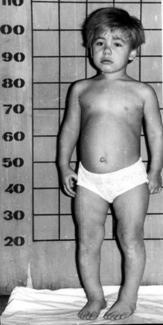
I тип — синдром Гурлер, синдром Шейе, синдром Гурлер-Шейе. Обусловлен дефицитом альфа-L-идуронидазы (фермент катаболизма мукополисахаридов). Заболевание постепенно приводит к накоплению в тканях гепарансульфата и дерматансульфата. Выделяют три фенотипа: синдром Гурлер, синдром Шейе и синдром Гурлер-Шейе.
Клиническая картина:
-
помутнение роговицы;
-
гаргоилизм: крупный череп, крутой лоб, запавшая переносица, толстые губы, большой язык, характерное выражение лица;
-
избыточный рост волос на теле (гипертрихоз, гирсутизм);
-
характерные деформации кистей и стоп: пальцы короткие, одинаковые по длине (изодактилия) расходятся веерообразно, напоминая трезубец;
-
тугоподвижность суставов.
-
короткое тело и шея
-
большой живот
-
возможна УО
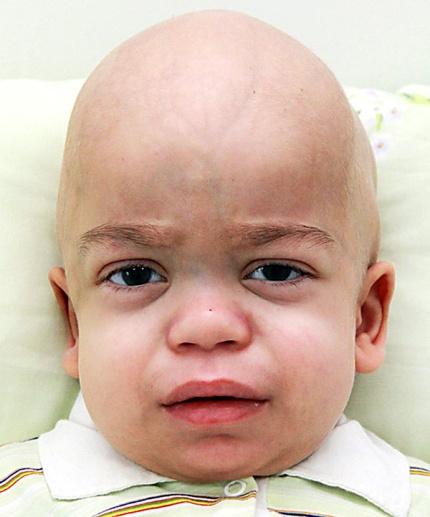
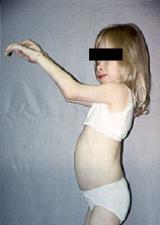
II тип — синдром Хантера, одна из форм мукополисахаридоза, мукополисахаридоза 2-го типа (MPS II), редкое рецессивное Х‑сцепленное генетическое заболевание из группы лизосомных болезней накопления. Возникает в результате дефицита ряда ферментов, что приводит к накоплению белково‑углеводных комплексов и жиров в клетках.
Клиническая картина:
-
утолщение ноздрей, губ, языка,
-
тугоподвижность суставов,
-
задержка роста
-
шумное дыхание, повторные риниты, паховые и пупочные грыжи.
-
грубые черты лица (гаргоилизм),
-
низкий грубый голос,
-
возникают частые острые респираторные вирусные инфекции.
-
утолщенная кожа,
-
короткая шея,
-
редкие зубы.
-
возможна УО
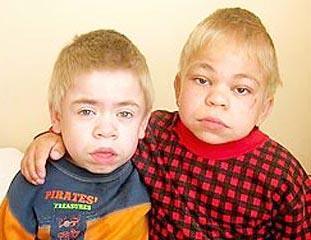
III тип — синдром Санфилиппо, группа клинически сходных редких наследственных заболеваний, относящихся к лизосомным болезням накопления. Заболевания связаны с дефицитом того или иного фермента, в результате чего в лизосомах накапливается один тип гликозаминогликанов — гепарансульфат. Нарушения, в соответствии с дефектом гена (дефицитом того или иного фермента), разделяют на четыре формы.
Клиническая картина:
-
задержка развития
-
гиперактивность
-
беспокойность
-
в определённом возрасте утрачивают все приобретённые навыки, развивается УО
-
гипертрихоз
-
реже возникают проблемы с ушами, лёгкими и т.д.
-
уплощенная переносица
-
макроглоссия
-
зубы плохо сформированы
-
живот выпячен
-
утолщенная кожа