

Micro-Nano Technology for Genomics and Proteomics BioMEMs - Ozkan
.pdf122 |
RONALD PETHIG |
[49] or of 20 N/m2 for T cells [13]. Plant cells, because of their comparatively large size and the presence of large vacuoles, are more sensitive to shear stress (either from fluid flow or DEP forces) than mammalian cells [52].
It is well known that cell membranes can be disrupted by forced oscillation at frequencies greater than around 10 kHz, and this is the basis for using high-power sonication to disintegrate cells. Cell-destruction by DEP is primarily related to a field-induced breakdown of the physical integrity of the plasma membrane, as evidenced by the fact that the internal structure of the cell appears (under phase-contrast microscopy) to remain intact for some time and is even manipulable by DEP (e.g., [24]). Controlling the reversible electrical breakdown of membranes is an essential tool used in electroporation and electrofusion of cells (e.g., [63]). Electroporation (also called electropermeabilization) is typically achieved by subjecting the cells to a voltage pulse, where the field can range from around 5 MV/m for microsecond pulses, down to 0.1 MV/m for millisecond pulses, depending on the cell type. To obtain a significant DEP force, the factor (E. )E in eqn. 4.3 should be of the order 1013 V2/m3, which corresponds to an applied field less than 10 kV/m [69]. The reason a relatively weak DEP field can cause an electrical breakdown of the plasma membrane is because the field is ‘amplified’ in the form of a potential induced across the membrane. For a model, spherical, cell the frequency dependence of the field Em acting across the membrane is given ([67], and references cited therein) by:
Em (ω) = |
1.5(r/d)E cos θ |
(4.10) |
|||
(1 |
+ |
ω2 |
τ 2)1/2 |
||
|
|
|
|
|
|
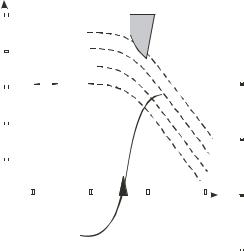
In eqn. 4.10 the factor r /d is the ratio of the cell radius r to the membrane thickness d, and τ is the relaxation time given by eqn. 4.4. Values for the membrane thickness are variously cited in the literature as being in the range 3–10 nm, so that the factor r /d can have a value of the order 103 and greater. Eqn. 4.10 therefore predicts that at low frequencies the field Em acting across a mammalian cell membrane can exceed the applied field E by a factor of at least 103. Polarizations associated with the electrical double layer at the membrane surface are not included in eqn. 4.10, and so we can expect even larger values for Em at the lowest frequencies. The greatest field stress is created across the membrane region that lies in a radial direction parallel with the field (θ = 0). At high frequencies (ωτ 1) the value for Em falls to low values. The estimated frequency dependence of the membrane field Em for T cells (Jurkat E6-1: ATCC, TIB-152) is shown in figure 4.13 for a range of values of (E. )E commonly obtained in DEP experiments using microelectrodes and applied voltages up to 20 V r.m.s. Also shown in figure 4.13 is the DEP spectrum for the T cells and the fxo value obtained with a medium conductivity of 40 mS/m [74], together with the frequency-field ‘window’ within which cell bursting has been observed (unpublished work). These results, together with observations for other cells (e.g., human blood cells; Bristol-8 B lymphoblastoid cells [ECACC 85011436]; HL-60 promyelocytic leukemia cells [ECACC 98070106]; and BeWo trophoblasts [ECACC 86082803]) suggest that the onset of irreversible damage (and cell bursting) can be induced using moderate to high values of a positive DEP force at frequencies where the field stress across the membrane approaches a value of 108 V/m or higher. Cancer cells appear to be more fragile than normal cells, and this is probably related to the fact they tend to have larger r/d values and therefore higher values of induced field stress Em . Under standard experimental
CELL PHYSIOMETRY TOOLS BASED ON DIELECTROPHORESIS
T Cells (Jurkat E6-1)
|
9 |
|
|
|
|
|
1016 |
|
|
|
|
|
|
|
|
|
|
FieldFrequency |
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Window for Cell |
||||
|
8 |
|
|
|
|
|
|
1015 |
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Bursting |
|||||
|
|
|
|
|
|
1014 |
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
(V/m) |
7 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1.0 |
|
|
||
|
|
|
(E.grad)E = 1013 (V2/m3) |
|
|
|
|
|
|
|
|
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
m |
|
|
|
|
|
|
|
|
|
|
|
-1 |
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
E |
6 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(2πτ) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
fxo |
|
DEP |
0.5 |
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
0 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
2 |
|
|
|
|
|
|
4 |
6 |
8 |
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Log Frequency (Hz) |
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
-0.5 |
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
123
Normalized DEP response
FIGURE 4.13. The frequency range where bursting (of more than 5% of total cell population) of T cells (Jurkat E6-1) occurs is plotted against the estimated induced membrane field stress Em, derived using eqn. 4.10, for various values of the parameter (E. )E of eqn. 4.3. Also shown is the DEP spectrum for the cells, the spread of fxo values, and the characteristic frequency corresponding to (2π τ )−1. (Talery, Lee & Pethig, unpublished)
conditions, the Em values experienced by cancer cells can also be larger because of their larger membrane capacitance values, associated with ‘rougher’ membrane surfaces than normal cells, which lowers the frequency where positive DEP occurs. The situation is more complicated for TWD experiments, because the bursting of a few cells can often be observed [77] at frequencies where the so-called FUN effect occurs [34]. This is characterized by unstable motions that can include cell rotation as well as rapid reversals of a cell’s direction of TWD motion. Significant shear forces can be exerted on some types of cells during such behavior, and this will contribute to the onset of field-induced cell destruction.
CONCLUDING COMMENTS
DEP is worthy of being considered a competitive alternative to the more conventional methods of cell concentration and separation, such as centrifugation, filtration, fluorescence activated cell sorting, or optical tweezers. Because DEP can operate directly on native, unlabeled cells, it eliminates the expense, labor and time of labeling and tagging, as well as the development and validation of such labels and tags. The same basic DEP method has the (probably unique) capability of isolating and analyzing a wide range of particle types (cells, bacteria, viruses, DNA and proteins) using one basic procedure.
After their selective isolation and recovery by DEP, cells remain viable for further analysis, processing or cell therapy. Multiple parameters on individual live cells can be determined and, if desired, specific cell types can be collected. Because it utilizes electronic signals, the technology is capable of extensive automation, is inexpensive and portable. DEP
124 |
RONALD PETHIG |
can operate under sterile conditions. Although the ability to selectively isolate cells without harming them is important for many perceived applications, there are also advantages to be gained by being able to destroy selected target cells. DEP appears capable of achieving this objective. Just as there are limitations in the use of electrofusion and electroporation (mainly associated with the fact that not all cell types can be treated with the same ease, and with irreproducibility between different laboratories) we can expect that efficient and reproducible protocols for selective cell destruction by DEP will not be so easily achieved as the DEP isolation of viable target cells. But it is a worthwhile challenge.
REFERENCES
[1]M. Abraham. The Classical Theory of Electricity and Magnetism, English Translation of 8th German Ed., Hafner Publ. Co., New York, 1932.
[2]W.M. Arnold, H.P. Schwan, and U. Zimmermann. Phys. Chem., 91:5093, 1987.
[3]D.J. Bakewell, I. Ermolina, H. Morgan, J.J. Milner, and Y. Feldman. Biochim. Biophys. Acta., 1493:151, 2000.
[4]J.B. Bates, T.T. Chu, and W.T. Stribling. Phys. Rev. Lett., 60:627, 1988.
[5]J. Bauer. In J. Bauer (ed.), Cell Electrophoresis CRC Press, Boca Raton, 1994.
[6]F.F. Becker, X.B. Wang, Y. Huang, R. Pethig, J. Vykoukal, and P.R.C. Gascoyne. J. Phys. D: Appl. Phys., 27:2659, 1994.
[7]F.F. Becker, P.R.C. Gascoyne, Y. Huang, and X.B. Wang. US patent 5 993 630, 1999.
[8]D.J. Bennett, B. Khusid, C.D. James, P.C. Galambos, M. Okandan, D. Jacqmin, and A. Acrivos. Appl. Phys. Lett., 83:4866, 2003.
[9]J.P.H. Burt, T.A.K. Al-Ameen, and R. Pethig. J. Phys. E: Sci. Instrum., 22:952, 1989.
[10]J.P.H. Burt, R. Pethig, P.R.C. Gascoyne, and F.F. Becker. Biochim. Biophys. Acta, 1034:93, 1990.
[11]J.P.H. Burt, R. Pethig, and M.S. Talary. Trans. Inst. MC, 20:82, 1998.
[12]E.G. Cen, C. Dalton, Y. Li, S. Adamia, L. M. Pilarski, and K.V.I.S. Kaler. J. Microbiol. Meth., 58:387, 2004.
[13]K.K Chittur, L.V. McIntire, and R.R. Rich. Biotechnol. Prog., 4:89, 1988.
[14]K.S. Cole. Membranes, Ions and Impulses, Univ. of Calif. Press, 1972.
[15]E.B. Cummings. IEEE Eng. Med. Biol. Magazine, 22(6):75, 2003.
[16]C.W. Einolf and E.L. Carstensen. Biophys. J., 9:634, 1969.
[17]M. Fr´en´ea, S.P. Faure, B. Le Piouffle, Ph. Coquet, and H. Fujita. Mat. Sci. Eng. C, 23:597, 2003.
[18]H. Frohlich¨ . Theory of Dielectrics, Oxford University Press, Oxford, pp. 26–28, 1949.
[19]G. Fuhr, T. Muller,¨ Th. Schnelle, R. Hagedorn, A. Voigt, S. Fiedler, W. M. Arnold, U. Zimmermann, B. Wagner, and A. Heuberger. Naturwissenschaften, 81:528, 1994a.
[20]G. Fuhr, H. Glasser, T. Muller,¨ and T. Schnelle. Biochim. Biophys. Acta, 1201:353, 1994b.
[21]G. Fuhr, Th. Schnelle, R. Hagedorn, and S.G. Shirley. Cell. Eng., 1:47, 1995.
[22]G. Fuhr, C. Reichle, T. Muller,¨ K. Kahlke, K. Schutze, and M. Stuke, Appl. Phys., A69:611, 1999.
[23]P.R.C. Gascoyne, Y. Huang, R. Pethig, J. Vykoukal, and F.F. Becker. Meas. Sci. Technol. 3:439, 1992.
[24]P.R.C. Gascoyne, R. Pethig, J.P.H. Burt, and F.F. Becker. Biochim. Biophys. Acta, 1149:119, 1993.
[25]P.R.C. Gascoyne and J. Vykoukal. Electrophoresis 23:1973, 2002.
[26]A.D. Goater, J.P.H. Burt, and R. Pethig. J. Phys. D: Appl. Phys., 30:L65, 1997.
[27]D.C. Graham. Chem. Rev., 41:441, 1947.
[28]E.H. Grant, R.J. Sheppard, and G.P. South. Clarendon Press, Oxford, 1978.
[29]S. Grimnes and O.G. Martinsen. Bioimpedance and Bioelectricity, Academic Press, San Diego, 2000.
[30]R. Hagedorn, G. Fuhr, T. Muller,¨ and J. Gimsa. Electrophoresis, 13:49, 1992.
[31]J.B. Hasted. Aqueous Dielectrics, Chapman & Hall, London, 1973.
[32]K.F. Hoettges, M.P. Hughes, A. Cotton, N.A.E. Hopkins and M.B. McDonnell. IEEE Eng. Med. Biol. Magazine, 22(6):68, 2003.
[33]Y. Huang and R. Pethig. Meas. Sci. Technol., 2:1142, 1991.
[34]Y. Huang, J.A. Tame, and R. Pethig. J. Phys. D: Appl. Phys., 26:1528, 1993.
CELL PHYSIOMETRY TOOLS BASED ON DIELECTROPHORESIS |
125 |
[35]Y. Huang, X.-B. Wang, R. Holzel,¨ F.F. Becker, and P.R.C. Gascoyne. Phys. Med. Biol., 40:1789, 1995.
[36]Y. Huang, X.-B. Wang, F.F. Becker, and P.R.C. Gascoyne. Biochim. Biophys. Acta, 1282:76, 1996.
[37]Y. Huang, X.-B. Wang, F.F. Becker, and P.R.C. Gascoyne. Biophys. J., 73:1118, 1997.
[38]Y. Huang, X.-B. Wang, P.R.C. Gascoyne, and F.F. Becker. Biochim. Biophys. Acta, 1417:51, 1999.
[39]Y. Huang, K.L. Ewaituhon, M. Tirado, R. Haigis, A. Foster, D. Ackley, M.J. Heller, J.P. O’Connell, and M. Krihak. Anal. Chem., 73:1549, 2001.
[40]Y. Huang, S. Joo, M. Duhon, M. Heller, B. Wallace, and X. Xu. Anal. Chem., 74:3362, 2002.
[41]M.P. Hughes. Nanoelectromechanics in Engineering and Biology, CRC Press, Boca Raton, 2002,a.
[42]M.P. Hughes. Electrophoresis, 23:2569, 2002,b.
[43]M.P. Hughes, R. Pethig, and X.-B. Wang. J. Phys. D: Appl. Phys., 29:474, 1996.
[44]T. Inoue, R. Pethig, T.A.K. Al-Ameen, J.P.H. Burt, and J.A.R. Price. J. Electrostat., 21:215, 1988.
[45]A. Irimajiri, T. Hanai, and A. Inouye. J. Theor. Biol., 78:251, 1979.
[46]T.B. Jones and G.A. Kallio. J. Electrostatics, 6:207, 1979.
[47]T.B. Jones. Electromechanics of Particles, Cambridge University Press, Cambridge, 1995.
[48]H. Li and R. Bashir. Sens. Actu. B: Chem., 86:215, 2002.
[49]L.B. Leverett, L.D. Hellums, C.P. Alfrey, and E.C. Lynch. Biophys. J., 12:257, 1972.
[50]J. Lyklema. Fundamentals of Interface and Colloid Science, Vol. 2: Solid-Liquid Interfaces, Academic Press, San Diego, 1995.
[51]N. Manaresi, A. Romani, G. Medora, L. Altomare, A. Leonardi, and M. Tartagni. IEEE J. Sol. St. Circ., 38:2297, 2003.
[52]G.H. Markx and R. Pethig. Biotechnol. & Boieng., 45:337, 1995.
[53]G.H. Markx, Y. Huang, X.F. Zhou, and R. Pethig. Microbiology, 140: 1994a.
[54]G.H. Markx, M.S. Talary, and R. Pethig. J. Biotechnology, 32:29, 1994b.
[55]S. Masuda, M. Washizu, and I. Kawabata. IEEE Trans. Ind. Appl., 24:214, 1988.
[56]J.C Maxwell. A Treatise on Electricity and Magnetism, 3rd Ed., Vol. 1, Ch. 9, Clarendon Press, Oxford, 1891.
[57]H. Morgan, N.G. Green, M.P. Hughes, W. Monaghan, W., and T.C. Tan. J. Micromech. Microeng., 7:65, 1997.
[58]H. Morgan, A.G. Izquierdo, D. Bakewell, N.G. Green, and A. Ramos. J. Phys. D: Appl. Phys., 34:1553, 2001.
[59]H. Morgan and N.G. Green. AC Electrokinetics: Colloids and Nanoparticles, Research Studies Press, Baldock, 2003.
[60]J.R. Morgan and R. Pethig. J. Phys. E: Sci. Instrum., 12:1132, 1979.
[61]T. Muller,¨ A. Pfennig, P. Klein, G. Gradl, M. J¨ager, and T. Schnelle. IEEE Eng. Med. Biol. Magazine, 22(6):51, 2003.
[62]T. Nagahama, N. Muramatsu, H. Ohshima, and T. Kondo. Colloid. Surf., 67:61, 1992.
[63]E. Neumann, A.E. Sowers, and C.A. Jordan. (Eds.) Electroporation and Electrofusion in Cell Biology, Plenum Press, New York, 1989.
[64]R. Paul, K.V.I.S. Kaler, and T.B. Jones. J. Phys. Chem., 97:4745, 1993.
[65]S.J. Paddison, R. Paul, and K.V.I.S. Kaler. J. Colloid Interface Sci., 183:78, 1996.
[66]R. Pethig. Dielectric and Electronic Properties of Biological Materials, J. Wiley, Chichester, 1979.
[67]R. Pethig. In I. Karube (ed.), Automation in Biotechnology, Elsevier Science Publishers B.V., 159–185, 1991.
[68]R Pethig. Crit. Rev. Biotechnology, 16:331, 1996.
[69]R. Pethig and G.H. Markx. Trends in Biotechnology, 15:426, 1997.
[70]R. Pethig and J.P.H. Burt. US Patent 5 795 457, 1998.
[71]R. Pethig and G.H. Markx. US Patent 5 814,200, 1998.
[72]R. Pethig, J.P.H. Burt, A. Parton, N. Rizvi, M.S. Talary, and J.A. Tame. J. Micromech. Microeng., 8:57, 1998.
[73]R. Pethig and J.P.H. Burt. European Patent EP 0 898 493, 2000.
[74]R. Pethig, V. Bressler, C. Carswell-Crumpton, Y. Chen, L. Foster-Haje, M.E. Garicia-Ojeda, R.S. Lee, G.M. Lock, M.S. Talary, and K.M. Tate. Electrophoresis, 23:2057, 2002.
[75]R. Pethig, M.S. Talary, R.S. Lee, B. Kusler and C. Carswell-Crumpton. In M.A. Northrup, K.F. Jensen and D.J. Harrison (eds.), Proc. µTAS 2003, The Transducers Research Foundation, ISBN: 0-9743611-0-0) pp. 1065–1068, 2003.
126 |
RONALD PETHIG |
[76]R. Pethig, M.S. Talary, and R.S. Lee. IEEE Eng. Med. Biol. Magazine, 22(6):43, 2003.
[77]R. Pethig, R.S. Lee, and M.S. Talary. JALA, 9:324, 2004.
[78]H.A. Pohl. J. Appl. Phys., 22:869, 1951.
[79]H.A. Pohl. Dielectrophoresis, Cambridge University Press, Cambridge, 1978.
[80]H.A. Pohl and R. Pethig. J. Phys. E: Sci. Instrum., 10:190, 1977.
[81]J.A.R. Price, J.P.H. Burt, and R. Pethig. Biochim. Biophys. Acta, 964:221, 1988.
[82]A. Ramos, H. Morgan, N.G. Green, and A. Castellanos. J. Phys. D: Appl. Phys., 31:2338, 1998.
[83]K. Ratanachoo, P.R.C. Gascoyne, and M. Ruchirawat. Biochim. Biophys. Acta, 1564:449, 2002.
[84]J. Rousselet, G. H. Markx, and R. Pethig. Colloids Surf. A: Physicochem. Eng. Aspects, 140:209, 1998.
[85]B. Saif, R.K. Mohr, C.J. Montrose, and T.A. Litovitz. Biopoymers, 31:1171, 1991.
[86]M. Sancho, G. Martinez, and C. Martin. J. Electrostat., 57:143, 2003.
[87]F.A. Sauer. In A. Chiabrera, C. Nicolini, and H. P. Schwan (eds.), Interactions between Electromagnetic Fields and Cells, Plenum Publ., New York, pp. 181–202, 1985.
[88]T. Schnelle, T. Muller,¨ and G. Fuhr. J. Electrostat., 50:17, 2000.
[89]H.P. Schwan. In A. Chiabrera, C. Nicolini, and H. P. Schwan (eds.), Interactions between Electromagnetic Fields and Cells, Plenum Publ., New York, pp. 75–97, 1985.
[90]J.W. Smith. Electric Dipole Moments, Butterworth Scientific Publ., London, 7–11, 1955.
[91]M. Stephens, M. Talary, R. Pethig, A.K. Burnett, and K.I. Mills. Bone Marrow Transplant 18:777, 1996.
[92]J. Suehiro and R. Pethig. J. Phys. D: Appl. Phys., 31:3298, 1998.
[93]J. Suehiro, R. Hamada, D. Noutomi, M. Shutou, and M. Hara. J. Electrostatics, 57:157, 2003a.
[94]J. Suehiro, D. Noutomi, M. Shutou, and M. Hara. J. Electrostatics, 58:229, 2003b.
[95]V.L. Sukhorukov, G. Meedt, M. Kurschner, and U. Zimmermann. J. Electrostat., 50:191, 2001.
[96]S. Takashima. Electrical Properties of Biopolymers and Membranes, Adam Hilger, Bristol, 1989.
[97]M.S. Talary and R. Pethig. IEEE Proc. Sci. Meas. Technol., 141:395, 1994.
[98]M.S. Talary, J.P.H. Burt, J.A. Tame, and R. Pethig. J. Phys. D: Appl. Phys., 29:2198, 1996.
[99]J. Voldman, M. Toner, M.L. Gray, and M.A. Schmidt. J. Electrostat., 57:69, 2003.
[100]K.W. Wagner. Archiv. Elektrotechnik, 2:S371, 1914.
[101]X.B. Wang, R. Pethig, and T.B. Jones. J. Phys. D: Appl. Phys., 25:905, 1992.
[102]X.B. Wang, Y. Huang, J.P.H. Burt, G.H. Markx, and R. Pethig. J. Phys. D: Appl. Phys., 26:312, 1993a.
[103]X.B. Wang, Y. Huang, R. Holzel,¨ J.P.H. Burt, and R. Pethig. J. Phys. D: Appl. Phys., 26:1278, 1993b.
[104]X.B. Wang, Y. Huang, P.R.C. Gascoyne, F.F. Becker, R. Holzel,¨ and R. Pethig. Biochim. Biophys. Acta, 1193:330, 1994a.
[105]X.B. Wang, Y. Huang, F.F. Becker, and P.R.C. Gascoyne. J Phys. D: Appl. Phys., 27:1571, 1994b.
[106]X-B. Wang, Y. Huang, X. Wang, F.F. Becker, and P.R.C. Gascoyne. Biophysical J., 72:1887, 1997.
[107]X.J. Wang, F.F. Becker, and P.R.C. Gascoyne. Biochim. Biophys. Acta, 1564:412, 2002.
[108]M. Washizu and O. Kurosawa. IEEE Trans. Ind. Appl., 26:1165, 1990.
[109]M. Washizu, S. Suzuki, O. Kurosawa, T. Nishizaka, and T. Shinohara. IEEE Trans. Ind. Appl., 30:835, 1994.
[110]J. Yang, Y. Huang, X. Wang, X. B. Wang, F.F. Becker, and P.R.C. Gascoyne. Biophys. J., 76:3307, 1999.
5
Hitting the Spot: The Promise
of Protein Microarrays
Joanna S. Albala
Biology & Biotechnology Research Program, Lawrence Livermore National Laboratory, 7000 East Avenue, L-448, Livermore, CA 94550
5.1. INTRODUCTION
With the thrust of scientific endeavor moving from genomics to proteomics, the protein array provides a powerful means by which to examine hundreds to thousands of proteins in parallel. A result of the many genome projects has been the advance of automation and robotic procedures to manipulate biomolecules using a high-throughput, systematic approach. The promise of the protein microarray is the ability to interrogate a large number of proteins simultaneously in a high-density format for disease diagnosis, prognosis or efficacy of therapeutic regime as well as for biochemical analysis. Similar to a DNA microarray, each spot on a protein array can be identified based on its addressability on the planar surface.
Protein microarrays can be generally classified into two categories: 1) protein profiling arrays and 2) functional protein arrays. Protein profiling arrays typically are arrays of antibodies used to compare two different cell types or disease states for novel biomarker identification, protein abundance or alteration [1] whereas functional protein arrays are usually comprised of recombinant proteins used to identify protein-protein, protein-DNA, or protein-small molecule interactions or other biochemical activities [2] and for review see [3–5]. Protein arrays have been designed to encompass the protein equivalent of whole organisms [6] and have been used for cell and tissue-based analyses as well [7, 8].
The concept of proteomic analysis by protein array has been emerging from both academic and industrial laboratories worldwide with increasing success. The generation of new technologies to manipulate the diversity of cellular proteins for protein array applications is advancing the ability to work with this diverse population of biomolecules as well as
128 |
JOANNA S. ALBALA |
furthering a greater understanding of many different organisms and disease states. This review aims to provide a progress report of protein microarray advances to date and provides an example for the use of protein microarrays to identify protein-protein interactions in homologous recombination and DNA repair pathways.
5.2. GENERATION OF PROTEIN MICROARRAYS
There are many variables to consider in developing a protein array. These include content, surface chemistry, array production, and detection. Each of these will be discussed in turn below.
5.2.1. Content
Content has proven to be the greatest bottleneck for the generation of protein and antibody arrays. Although large collections of cDNAs are available from the I.M.A.G.E. cDNA Consortium at Lawrence Livermore National Laboratory [9], Mammalian Gene Collection from the National Cancer Institute [10] and the FLEXgene collection at the Harvard Institute of Proteomics [11], it is still a large undertaking to produce proteins in high-throughput.
5.2.1.1.Proteins Most efforts have focused on generating recombinant protein using E. coli for its high productivity and ease of use [12]. High-throughput protein production using E. coli has been successful for proteins of several species from bacteria to man [13–16]. The majority of these studies have focused on the production of a 96-well plate of recombinant proteins and have applied both traditional as well as cell-free expression from bacteria [17–19]. Furthermore, several studies have successfully applied these techniques for the production of large numbers of proteins for high-density protein arrays [20]. Synder and co-workers have identified several novel kinases and lipid-binding proteins by generating a functional protein array encompassing nearly the entire proteome of Saccharomeyces cerevisiae [6]. Most recently, potential autoimmune disease-associated biomarkers were identified using a high-density array of 2400 human proteins devised by robotic spotting of the recombinant proteins onto glass microscope slides pretreated with a polyacrylamide coating [21].
5.2.1.2.Antibodies The successful generation of protein arrays for protein profiling in a manner similar to that of a DNA microarray requires a different type of content for the generation of the array, the capture agent. Most protein arrays for this purpose have employed monoclonal antibodies due to their high specificity and affinity for the target from which they were generated; however, polyclonal antibodies have also been used successfully [22]. The development of monoclonal or polyclonal antibodies is a laborious and costly process. As a result, several other capture agents have been described for use in the development of protein microarrays. Fab/scFv fragments from phage display libraries have been used for protein profiling [23]. Phage-display libraries may allow for the production of a large and diverse population of capture agents on a proteomic scale more rapidly than is possible by immunization-based methods of antibody production [24]. In addition, in vitro-based
HITTING THE SPOT: THE PROMISE OF PROTEIN MICROARRAYS |
129 |
methods of ribosome or mRNA display that physically links the capture agent to its cognate mRNA have been used successfully for protein microarray applications [25]. Aptamer arrays have also been demonstrated [26]. Aptamers are short stretches of DNA or RNA that are generated from an in vitro process termed “SELEX” for systematic evolution of ligands by exponential enrichment. It is possible to use this process to generate aptamers with high binding affinities to specific ligands including proteins [27]. It is noteworthy that the high-throughput production of capture agents relies on the successful generation of target proteins resulting in both the generation of the antibody arrays as well as for validation of the affinity, specificity, and cross-reactivity of each capture agent on the array.
5.2.2. Surface Chemistry
Protein arrays have been generated on PVDF [28], nitrocellulose [20], plastic [2] or more commonly on glass microscope slides [29] which are compatible with standard robotic arrayers and detection instrumentation used for printing and analyzing DNA microarrays. The surface to which proteins are bound is key in the development of protein microarrays and certain criteria must be considered. First, the surface needs to be advantageous for protein binding; second, it must be amenable to high-throughput production and application; third, low auto fluorescence of the surface is required as fluorescence is the most common means of detection using protein microarrays; and finally, the surface must strive to provide an environment suitable to maintain the native structure of the proteins or capture agents [30]. Several different surface coatings have been used for generating protein microarrays including glass slides activated with aldehyde [29], poly-lysine [31] or a homofunctional crosslinker [32], agarose [33], polyacrylamide gel pads [34], hydrogel [35]. Two recent reports have compared the use of standard surface chemistry with a hydrogel polyacrylamide support and found the hydrogel surface to have better sensitivity with lower background, thus proving to be a superior surface from which to produce protein arrays [30, 35]. Nitrocellulose-coated slides have also been employed for several protein array applications [20, 36].
Efforts have been made to orient proteins on the array to facilitate protein-protein interactions. For example, nickel-coated slides have been used for the generation of protein arrays of recombinant his-tagged proteins [6]. Protein G-coated and strepavidin-coated slides have been utilized to orient antibodies for the generation of antibody microarrays [37, 38], as have microfabricated pillars [39] and repeated application of the target and capture agent to the same address on the array to direct the application of the protein content onto the array surface [22].
5.2.3. Microarray Production
Protein microarray production requires careful preparation of samples and slides prior to printing, and attention to the humidity, temperature, and dust levels in the printing environment [5]. The implementation of automated protein array production includes deposition by pin-based or microdispensing liquid-handling robots [40], photolithography [41, 42], or ink-jet printing technology [43]. Piezoelectric dispensing of proteins from borosilicate glass capillaries is a common method of protein array production that deposits samples onto a planar surface by non-contact printing. It has been demonstrated that addition of bovine
130 |
JOANNA S. ALBALA |
serum albumin (BSA) to a low ionic strength buffer can effectively minimize nonspecific protein adsorption to the glass capillaries, a common drawback of this approach [44].
Another new approach is affinity contact printing whereby different proteins are attached to three-dimensional features on an array that can then capture other proteins from a complex mixture. These 3D features can then be used to stamp the captured proteins onto another surface for further analysis by mass spectrometry [45].
5.2.4. Detection
There are several methods for detection of protein interactions using an array format. The most common rely on fluorescently-labeled antibodies or proteins applied to the array. A typical protein profiling experiment will employ an ELISA-based sandwich approach where antibodies are arrayed onto the slide, sample is applied, followed by addition of another capture antibody which is either directly labeled with a fluorochrome (direct detection) or a labeled secondary (indirect detection) is applied [46]. Alternatively, the sample material can be directly labeled and compared to a control sample population that has been labeled with a different fluorophore [1]. Other detection methods less commonly used include SPR (surface plasmon resonance) or RCA (rolling circle amplification). SPR relies on changes in the angle of reflection when a protein interaction occurs on the surface of a gold-coated chip or array and the light caused by the binding of the probe to the immobilized protein is measured. An advantage of SPR is the use of unlabeled probes; however, this method is less sensitive than methods using fluorescence, chemiluminescence or radioactivity for detection (for review see [5]). RCA employs a sandwich immunoassay in which the detection antibodies are linked to DNA sequences that can be amplified and hybridized with complimentary fluorescentlylabeled oligonucleotides to detect the interaction between the antibody and its target [47]. Schweitzer et al. in a study of 51 cytokines from cultured dendritic cells demonstrated the differential regulation of several cytokines at biologically relevant concentrations [48].
5.3. PROTEIN ARRAYS FOR ANALYSIS OF PROTEINS INVOLVED IN RECOMBINATION & DNA REPAIR
5.3.1. Protein Expression Microarrays
We have developed protein expression microarrays for the production of recombinant human proteins using a baculovirus-based approach in a 96-well format. Heterologous protein production of mammalian proteins in E. coli often results in improperly modified, misfolded or insoluble protein [12]. For these reasons, we have focused our efforts on heterologous production of human protein using the baculovirus expression system [49]. Our process of protein production begins with the selection of cDNA clones from the I.M.A.G.E. collection. We produce recombinant baculoviruses with a PCR-based strategy into a custom transfer vector modified to contain an N-terminal histidine tag for purification of recombinant proteins. Automation is employed to set up the molecular biological reactions needed for PCR amplification, cleanup and cloning. A database has been developed to track the process of protein production. All reactions are performed in a 96-well plate format for miniaturization of the baculovirus paradigm. A 1.5 ml, 96-well insect cell culture system

HITTING THE SPOT: THE PROMISE OF PROTEIN MICROARRAYS |
131 |
has been devised for the expression and purification of microgram amounts of protein that is ideal for protein microarray applications.
We have employed our miniaturized protein production strategy for the generation of protein fragments for mapping interactions among domains of critical DNA repair proteins in the homologous recombinational repair pathway [50] as well as for the development of protein interaction microarrays [51]. Rad51 is a central protein in the process of homologous recombinational repair (for review see [52, 53]). Several human proteins with homology to Rad51 have been identified. These proteins are known as the Rad51 paralogs and include Rad51B, Rad51C, Rad51D, Xrcc2 and Xrcc3 [54–57]. Recent studies have shown that the Rad51 paralogs exist in two complexes in vivo independent of Rad51, BCDX2 and CX3 [58–61]. We were interested in identifying the binding sites between the different Rad51 paralogs in the BCDX2 complex. To this end, we used computational modeling against the Pyroccocus furious crystal structure [62], to direct the generation of deletion mutants for the individual paralog proteins. Recombinant proteins for the deletion mutants of each of the Rad51 paralog proteins were produced using our miniaturized baculoviral expression system (i.e., protein expression microarrays). A western blot of the deletion mutant series is shown in Figure 5.1. These include the deletion mutants for Rad51B, 1–75 and 76–350, Rad51C, 1–79, 79–376, and Rad51D, 4–77, 77–379, as well as several point mutants of the Rad51B, Rad51C and Rad51D proteins. Using yeast two-hybrid and coimmunoprecipitation techniques, we have demonstrated that a deletion construct of Rad51B containing amino acids residues 1–75 interacts with the C-terminus of Rad51C amino acids
Rad51B |
Rad51B 1-75 |
Rad51B 76-350 |
Rad51B K114A |
Rad51B K114R |
Rad51B K114A |
Rad51B K114R |
Rad51C |
Rad51C 1-79 |
Rad51C79-376 |
Rad51C K131A |
Rad51C K131R |
|
|
|
mRad51D |
mRad51D 4-77 |
mRad51D 77-328 |
mRad51D S111T |
mRad51D K113A |
mRad51D K114R |
|
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
|
|
|
13 |
14 |
15 |
16 |
17 |
18 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
50 |
|
|
|
|
|
|
|
50 |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
37.5 |
|
|
|
|
|
|
|
37.5 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
25 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
20 |
|
|
|
|
|
|
|
|
|
|
|
|
|
25 |
|
|
|
|
|
|
|
15 |
||
|
|
|
|
|
|
|
|
|
|
|
|
20 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
15 |
|
|
|
|
|
|
|
|
|
|
FIGURE 5.1. Rad51 paralog proteins produced using a miniaturized protein expression system in baculovirus (protein expression microarrays). Lane 1: Full-length Rad51B, Lanes 2–3: Deletion mutants of Rad51B, Lanes 4–7: Point mutants in the ATP binding site of Rad51B, Lane 8: Full-length Rad51C, Lanes 9–10: Deletion mutants of Rad51C; Lanes 11–12: Point mutants in the ATP binding site of Rad51C, Lane 13: Full-length Rad51D, Lanes 14–15: Deletion mutants of Rad51D, Lanes 16–18: Point mutants in the ATP binding site of Rad51D. Blots were incubated with a monoclonal antibody to an N-terminal his-tag (Sigma) and proteins were visualized using enhanced chemiluminescence (ECL, Amersham). Protein standards are shown representing molecular weight in kilodaltons (kDa).