

nevra_esche_ekzamen
.pdfкомбинированное лечение. При синдроме Веста применяют вигабатрин и тетракозактид. Препаратами второй очереди выступают вальпроаты. Если туберозный склероз протекает с парциальными эпиприступами, то базовой терапией считается сочетание вальпроатов с карбамазепином. При отсутствии эффекта в эту схему лечения включают ламотриджин. При генерализованных эпиприступах и парциальных пароксизмах в качестве монопрепарата и в комбинации с другими противоэпилептическими средствами могут применяться современные антиконвульсанты топирамат и леветирацетам.
•Терапия олигофрении проводится преимущественно путем нейропсихологической коррекции и комплексного психологического сопровождения ребенка. Назначение ноотропов и прочих стимулирующих нейропрепаратов противопоказано из-за наличия эписиндрома. При выявлении астроцитомы проводится динамическое наблюдение. Хирургическое удаление внутримозговой опухоли показано только при резком увеличении ее размеров с подъемом внутричерепного давления. Операцию проводят нейрохирурги.
•В отношении новообразований соматических органов применяется преимущественно выжидательная тактика. Хирургическое лечение проводится по показаниям, в основном в случаях, когда опухоль вызывает существенную дисфункцию органа или имеется угроза ее злокачественного течения.
Прогноз:
Прогноз для выздоровления неблагоприятный. При тяжелых системных изменениях высока летальность в детском и молодом возрасте от status epilepticus,сердечной,почечной или легочной недостаточности. Ведущей причиной болезненности и смертности при туберозном склерозе служат опухоли ЦНС,осложнения со стороны почек - вторая по значимости причина ранней смерти пациентов с туберозным склерозом.
124. Факоматозы. Нейрофиброматоз Реклингаузена. Клиника, диагностика, лечение, прогноз. Четверткова Нейрофиброматоз Реклингхаузена - заболевание, в основе - развитие опухолей (нейрофибром) из нервной ткани, как в ЦНС, так и на периферических нервах.
Симптомы зависят от локализации опухолей.
Ранние и типичные признаки - пигментные пятна размером более 1,5 см. Для постановки диагноза достаточно выявление минимум 6 пятен цвета «кофе с молоком» диаметром свыше 1,5 см Нейрофиброматозы являются наследственными заболеваниями, возникающими в связи с генными нарушениями.
Форма Реклингхаузена передается аутосомно-доминантно, вероятность рождения ребенка с патологией у больного родителя составляет 50%. Патологический ген на 17 хромосоме. В некоторых случаях нейрофиброматоз Реклингхаузена развивается как результат спонтанной мутации в семье, где случаи этого заболевания ранее не наблюдались.
Клиника разнообразна
Самый ранний признак - наличие у новорожденного или ребенка более 5-ти пигментных пятен диаметром 1,5 см и больше. Обычно они на туловище и шее, но м.б на лице и конечностях, имеют молочно-кофейную окраску, но могут встречаться фиолетовые, синие и даже депигментированные пятна.
Периферическая форма характеризуется развитием множественных неврином периферических нервов и нейрофибром в виде подкожных узелков. Опухоли могут локализоваться на туловище, шее, голове, конечностях. Они имеют округлую форму, подвижны и безболезненны при прощупывании. Размер - 1-2 см, но в отдельных случаях достигают значительной величины до 2 кг. Кожа над опухолями часто более пигментирована, на верхушке образования может отмечаться рост волос.
При появлении неврином на терминальных ветвях нервных стволов приводит к затруднению лимфооттока и развитию «слоновости» (лимфедемы), которая проявляется увеличением объема конечности, части лица, языка. Часто сочетается с аномалиями развития скелета: сколиозом, асимметрией черепа, незаращением дужек позвонков и др. Специфический признак - пятнышко на радужке - узелок Лиша - у 94% пациентов.
Диагностика. Наиболее часто диагностируется с 3х до 16 лет. Пигментные пятна различного размера встречаются у большинства детей - большое кол-во пятен (6 и более) и/или 2 нейрофибромы - поводом для консультации дерматолога
Если в семейном анамнезе есть предрасположенность к заболеванию - посещать офтальмолога -1 раз в год до 6 лет т.к. бессимптомная форма часто выявляется этим специалистом - 2 и более узелка Лиша - повод для беспокойства.
Для определения локализации опухолей - КТ и МРТ позвоночника, МРТ и КТ гол.мозга Диагностика аномалий скелета - путем рентгенографии позвоночника и черепа - при выявлении аномалий скелета - ортопед
-необходима консультация невролога
-Решение вопроса о хирургическом лечении - нейрохирург
Лечение:
нет эффективных способов. Пациентам проводится симптоматическая терапия.
При большом размере периферических опухолей, развитии неврологических нарушений, появлении признаков сдавления мозга - операции по удалению опухолей. Нейрофибромы могут быть удалены с косметической целью. При их озлокачествлении (3-5% случаев) удаление опухоли дополняется курсами химиотерапии и лучевой терапии.
Прогноз
- благоприятный, рак - в редких случаях.
Но при распространенности пат.процесса трудоспособность человека снижается - причина инвалидности. (нарушения интеллекта, затруднение в обучении письму и чтению, освоении наук. )
Так как данное заболевание является генетическим, профилактических мероприятий по его предупреждению не разработано. Рекомендуется родителям обнаружить заболевание как можно раньше. В медицине установлено, что около 60 % людей с данной патологией имеют не яркое проявление симптоматики в виде пигментных пятен и нескольких новообразований на кожном покрове, которые требуют проведения небольшого лечения.
Дети с такой болезнью остаются относительно здоровыми, но в будущем у них возможно развитие осложнений и негативных последствий. Потому важно вовремя диагностировать патологию, чтобы обеспечить ребенку счастливую жизнь.
125. Факоматозы. Энцефалотригеминальный ангиоматоз Штурге-Вебера. Атаксия-телеангиэктазия. Цереброретинальный ангиоматоз Гиппеля-Линдау. Клиника, диагностика, подходы к лечению. (ИВАНОВ)
Ангиоматоз Штурге-Вебера характеризуется ангиоматозом лица, сосудистой оболочки глаз и твердой мозговой оболочки. Основной клинический симптом - темно-розовое сосудистое пятно на одной стороне лица и головы, иногда распространяющееся на язык, губы, небо, глотку, дно ротовой полости, заднюю поверхность шеи. заболевание может сопровождаться экзофтальмом и эпилепсией.
Клиника: кожные изменения в виде ангиоматозного невуса локализуются на лице, обычно с одной стороны, но могут быть и двусторонними. Немус имеет цвет портвейна и выражен уже при рождении. он локализован в верхней и средней части лица, иногда распространяется на нижнюю часть лица, шею, грудную клетку, конечность, не изменятся в течение жизни.
Неврологические нарушения. Эпилептические приступы возникают у 75-90% пациентов. Приступы фокальные, часто по принципу джексоновского марша. у 90% пациентов на 1-м году жизни регистрируются инфантильные спазмы. Эпилептические приступы при синдроме Штурге-Вебера резистентны к противоэпилептической терапии. характерны постприступные парезы и параличи, которые регрессируют через несколько часов после приступа.
Глазные изменения характеризуются униили билатеральной глаукомой, ангиомой сосудистой оболочки глаза, гетерохромией радужной оболочки. очень редко ангиомы расположены во внутренних органах. Комбинация синдрома Штурге вебера и ангиоматоза внутренних органов выделяется некоторыми авторами в отдельный синдром Клиппеля -Треноне- Вебера.
Нейрорадиологические симптомы энцефалотригеминального ангиоматоза включают венозные ангиомы мягкой мозговой оболочки, расположенные над затылочной или теменно-затылочной областью, поражение церебральной гемисферы, расширение субэпендимальных вен и сосудов перивентрикулярной зоны, гемиатрофия полушарий, атрофическая редукция белого вещества, экстраинтрацеребральные ангиомы.
Диагностика: КТ и МРТ мозга. КТ более информативна для обнаружения интракраниальных кальцинатов. МРТ лучше визуализирует ангиомы, изменения белого вещества и сосудистых сплетений. Ангиография используется только для прехирургической диагностики тк очень инвазивна. При ЭЭГ выявляют межполушарную асимметрию и эпилептические паттерны - фокальные и генерализованные.
Лечение симптоматическое, оперативное и консервативное.
Консервативная терапия направлена на коррекцию нервно-психического статуса пациентов и купирование эпилептических приступов. целью оперативного лечения является устранение косметических дефектов и предотвращение повторных кровоизлияний.
Атаксия-телеангиэктазия (синдром Луи-Бар) -
Первые клинические проявления появляются в возрасте 1-З лет.
Характерны следующие симптомы: телеангиэктазии, особенно на склерах, прогрессирующие мозжечковые расстройства в виде атаксии, интенционного тремора, скандированной речи, нистагма. Постепенно присоединяются экстрапирамидные симптомы:гипомимия, гиперкинезы, монотонность, слабая модулированность речи, Часто больные отстают в умственном развитии.
Наряду с кожными и неврологическими проявлениями при синдроме Луи-Бар наблюдаются склонность к заболеванию легких и дыхательных путей, что может привести к бронхоэктазии, пневмосклерозу. Частые заболевания системы органов дыхания, вероятно, обусловлены дисгаммаглобулинемией. Синдром Луи-Бар относят к иммунодефицитным состояниям. Диагностика: ОАК - у 1/3 пациентов наблюдается снижение количества лимфоцитов. Исследование уровня иммуноглобулинов крови, которое выявляет значительное снижение IgA и IgE, в 10-12% случаев IgG.
Из инструментальных способов диагностики синдрома Луи-Бар могут применяться УЗИ тимуса, МРТ головного мозга, фарингоскопия, риноскопия, рентгенография легких. При помощи УЗИ диагностируется аплазия или гипоплазия тимуса. МРТ головного мозга выявляет атрофию мозжечка, расширение IV желудочка. Рентгенография легких необходима для диагностики очаговой или крупозной пневмонии, выявления очагов пневмосклероза и бронхоэктатических изменений. Для лечения симптома Луи-Бар, помимо симптоматической терапии, в последние годы применяют пересадку тимуса. Продлению жизни пациентов, имеющих синдром Луи-Бар, способствует иммунокоррегирующая терапия препаратами тимуса и гамма-глобулином, витаминотерапия в высоких дозировках и интенсивная терапия любого инфекционного
процесса. По показаниям применяют противовирусные препараты, антибиотики широкого спектра действия, противогрибковые средства, глюкокортикостероиды.
Лечение заключается в хирургическом удалении солидной части гемангиобластомы;
Цереброретинальный ангиоматоз Гиппеля-Линдау -
При обследовании наиболее отчетливые изменения обнаруживаются на глазном дне: ангиоматоз сетчатки, множественные аневризмы, кисты, дегенерация сетчатки. Оба глаза поражаются в 50% случаев. Помимо глазных изменений, Отмечаются симптомы поражения мозжечка -атаксия, нистагм, скандированная речъ, адиадохокинез. В мозжечке патоморфологически обнаруживаются множественные кисты. В неврологическом статусе выявляются также признаки внутричерепной гипертензии. Заболевание неуклонно прогрессирует. а также для лечения применяют дегидратационные средства.,
126. Нервно-мышечные заболевания. Классификация. Клиника, диагностика, подходы к лечению. Кикот НМЗ - гетерогенная группа болезней, в основе - генетически детерминированное поражение нервно-мышечного аппарата, обусловленное мутациями отдельного гена (1 или оба аллеля), т.е. клинические проявления возникают либо из-за отсутствия генетической информации, либо из-за ее дефекта
Классификация: |
Клиника: |
* По локализации поражения: (картинка для представления) |
Ведущие симптомы: мышечная слабость, утомляемость, |
|
часто гипотония и симметричная атрофия мышц, особенно |
|
в поздних стадиях развития болезни, “крыловидные |
|
лопатки”, вставание “лесенкой” (=приемы Говерса), осиная |
|
талия, “утиная” походка, контрактуры, сердечно-легочная |
|
недостаточность (при поражении межреберных мышц), |
|
отсутствие сухожильных рефлексов, псевдогипертрофия |
|
икроножных мышц («симптом перевернутой бутылки”). |
Диагностика:
•БХ крови: КФК, АСТ, АЛТ, ЛДГ повыш (при искл. Забол-й печени)
•Электромиография (опре уровня поражения: мышечный, спинальный, невральный)
•Мышечная биопсия (миодистрофии, врожд миопатии)
•ДНК-диагностика (ПЦР, секвенирование по Сангеру) - ищем дефектный ген
•МРТ мышц (селективные выпадения, атрофии)
•МРТ ГМ (врожденные формы миопатий)
•периферические нервы (невральные амиотрофии)
•мотонейроны спинного мозга (спинальные амиотрофии)
•мышцы (миодистрофии, структурные миопатии)
•нервно-мышечный синапс
Лечение:
1. Медикаментозная терапия:
-улучшение трофики мышц - АК (карнитин, лейцин,метионин, глутаминовая кислота), витамины E, A, B, C
-Аденозинтрифосфорная кислота, карбоксилаза, церебролизин, рибоксин фосфаден (улучшение трофики мышц)
-ГКС (преднизолон)
-увел. циркуляции крови и снабжение тканей О2 - никотиновая кислота, ксантинол, никошпан, компламин, теоникол
-улучшение нервно-мышечной проводимости - антихолинэстеразные (галантамин, оксазил, пиридостигмин, амиридин)
-легочно-сердечная недостаточность - бронхолитики, иАПФ и антиаритмические препараты
2.Реабилитация:
•ЛФК щадящая
•Физиотерапия (против развития контрактур и сколиоза) - озокеритовые и парафиновые аппликации, хвойные ванны
•Ортезы
•Вспомогательная вентиляция в случае ДН
3.Хирургическое лечение контрактур
4.Молекулярно-генетические методы лечения
•Клеточные трансплантации
•Восстановление мышечной мембраны+воздействие на кальциевые каналы
•Замещение дистрофина уротропином
•Экзон-скиппинг
127. Прогрессирующие мышечные дистрофии (ПМД). ПМД Дюшенна и Беккера. Клиника, диагностика, лечение.
ПМД - группа клинически полиморфных генетически детерминированных заболеваний, в основе которых лежат первичные прогрессирующие дегенеративные изменения в мышечных волокнах.
Общий клинический симптомокомплекс включает - нарастающую мышечную слабость - атрофии. Локализация - преимущественно в проксимальных отделах конечностей и поясах конечностей.
ПМД Дюшенна
Клиника:
•мышечная слабость и атрофии преимущественно в проксимальных отделах конечностей и поясах конечностей (с псевдогипертрофиями, мышечные контрактуры и сухожильные ретракции)
•Костно-суставные нарушения
•Сердечно-сосудистые расстройства
•Снижение интеллекта
•Эндокринные нарушения
Стадии:
I – преклиническая стадия - нет признаков болезни
II – клиническая стадия выражены все симптомы - но сохранена ходьба + псевдогипертрофии мышц, лордоз
III – последняя стадия с потерей ходьбы + выражены деформация позвоночника, контрактуры сгибательного характера, атрофии мышц Развитие:
1.Первые симптомы проявляются к 3-5 годам жизни – мышечная слабость и утомляемость.
2.Отчетливое прогрессирование симптомов особенно заметно к 7-8 годам: меняется походка по типу утиной, из-за мышечной слабости ограничивается самостоятельное передвижение больных. Видны «икры гнома».
3.Двигательная активность утрачивается к 14-15 годам.
4.После потери пациентами возможности самостоятельно передвигаться, у них формируются новые контрактуры - коленные, тазобедренные, локтевые, межфаланговые; искривление позвоночника.
5.Прогрессирующая сердечная и дыхательная недостаточность становятся причиной гибели пациентов в возрасте 15–25 лет.
Дети используют миопатические приемы Говерса: вставание "лесенкой», «взбирание по самому себе» Плечевой пояс – «крыловидные лопатки» Продольные мышцы спины – гиперлордоз Мышцы ПБС – лягушачий живот Косые м. живота - осиная талия
Костно-суставные нарушения: |
ССС-расстройства: |
• сколиоз |
• нарушения ритма |
• гиперлордоз |
• дисфункции миокарда желудочков |
• деформации грудной клетки (килевидная, воронковидная) |
• соответствующие изменение на ЭКГ |
• деформации стоп по типу «стопы Фридрейха». |
• кардиомиопатия |
• На рентгенограмме костей выявляются: |
|
– признаки атрофии диафизов длинных трубчатых костей |
|
– истончение кортикального слоя |
|
– сужение костно-суставного канала |
|
– диффузный остеопороз |
|
|
|
ПМД Беккера
Начало в 10-15 лет, но может развиваться и после 15-20 лет. Протекает гораздо мягче формы Дюшенна.
Больные с этой формой миодистрофии доживают до зрелого возраста.
Могут быть эндокринные нарушения – гинекомастия, снижение либидо, импотенция. Ретракции сухожилий и контрактуры менее выражены, чем при миодистрофии Дюшенна. Кардиомиопатия может отсутствовать.
Диагностика ПМД: |
Лечение ПМД: симптоматическое |
||
1. |
Клин. картина |
1. |
ГКС – замедляют прогрессирование мышечной слабости, |
2. |
Повышение в 50-100 раз активности КФК, ЛДГ в крови |
пульмо/кардио-протективный эффект., снижают риск |
|
3. |
Дегенерация мышц по данным биопсии, ЭМГ |
развития ортопедических осложнений. |
|
4. |
Молекулярно-генетический анализ |
2. |
Метаболическая терапия: витамины группы В, |
ЭМГ: |
левокарнитин, препараты Са2+, витамин D |
||
- Снижение средней длительности потенциалов |
3. |
Физиотерапевтические процедуры |
|
двигательных единиц (ПДЕ) на 4-5 % от нормы, |
4. |
Физическая активность - обязательно, по возможности |
|
- Сниж амплитуды (358,3 ± 17,7 мкВ (при норме 600 мкВ)), |
5. |
β-адреноблокаторы и ингибиторы АПФ для |
|
- Фазность потенциалов - 79,30 ± 5,98 % (норма до 5 %), |
предупреждения кардиомиопатии, диуретики при сердечной |
||
- Спонтанная активность в виде потенциалов фибрилляции |
недостаточности |
||
|
|
6. |
Респираторная поддержка |
|
|
|
|
128. Прогрессирующие мышечные дистрофии (ПМД). Поясно-конечностные формы ПМД (Эрба-Рота), лице-плече-лопаточная ПМД (Ландузи-Дежерина). Клиника, диагностика, лечение.
ПМД - группа клинически полиморфных генетически детерминированных заболеваний, в основе которых лежат первичные прогрессирующие дегенеративные изменения в мышечных волокнах.
Общий клинический симптомокомплекс включает - нарастающую мышечную слабость - атрофии. Локализация - преимущественно в проксимальных отделах конечностей и поясах конечностей.
ПМД Эрба-Рота
Относится к конечностно-поясным миодистрофиям Аутосомно-рецессивный тип наследования
•Начало 14-16 лет, реже в 5-10 лет
•Атрофии и мышечная слабость преобладают в проксимальных отделах нижних конечностей и тазового пояса (иногда одновременно вовлекаются проксимальные отделы и пояса верхних конечностей. Позже присоединяется поражение мышц спины и дистальных отделов конечностей) • Псевдогипертрофии, контрактуры суставов и ретракции сухожилий практически не выражены
•Кардиальная патология (очень редко)
Лечение:
1. Медикаментозная терапия
–Препараты, улучшающие тканевой метаболизм (АТФ, коэнзимQ, рибоксин, актовегин, кортексин); преднизолон и дефлазакорт – при первых признаках
–Аминокислотные комплексы (церебролизин, глицин, метионин, глутаминовая, фолиевая кислоты).
2.Физиотерапия (обертывания и электрофорез с ферментными препаратами)
3.Лечебная физкультура
4.Ортопедическая коррекция
5.Для профилактики остеопороза показано назначение препаратов, содержащих витамин D3 и кальций
6.Наблюдение и лечение у кардиолога
Диагностика:
1.Характерная клиника
2.Мол-ген анализ
3.Повышение уровня КФК (в 5 раз) – начальный период
4.РГ ГК: расширение границ сердца, воспаление лёгочной ткани
5.ЭКГ: аритмии
6.УЗИ сердца: кардиомиопатии
Лечение – симптоматическое:
1.АТФ, витамины Е и группы В, тиоктовая кислота
2.ЛФК, физиотерапии
3.При кардиомиопатиях - инозин, сердечные гликозиды, антиаритмики
4.Ортопедическое лечение
5.ИВЛ при атрофии дыхательных мышц
ПМД Ландузи - Дежерина
Относится к лице-лопаточно-плечевой форме Аутосомно-доминантный тип наследования
Дебют заболевания в возрасте 10-20 лет с медленным прогрессированием
Клиника: |
Диагностика ЛЛПМД: |
|
• Гипомимия лица |
1. |
Клиническая картина |
• «Полированный» лоб |
2. |
Повышение уровня КФК (в 5 раз) |
• Слабость круговых мышц глаз (лагофтальм, симптом |
3. |
Мышечный тип поражения на ЭНМГ |
ресниц) |
4. |
Биопсия – дегенерация мышц |
• Слабость круговой мышцы рта (трудно надувать шарики, |
5. |
Мол-ген анализ |
свистеть или пить через соломинку, «Улыбка Джаконды», |
|
|
«поперечная улыбка», «губы тапира») |
Лечение – симптоматическое: |
|
• Трудности с поднятием и удержанием рук над головой |
1. |
Физиотерапия неэффективна в отношении повышения |
• «Крыловидные» лопатки |
мышечной силы или замедления прогрессирования |
|
• Широкий межлопаточный промежуто |
мышечной слабости и атрофии |
|
• Сколиоз |
2. |
Ортопедическая коррекция, ЛФК |
• Уплощение грудной клетки |
3. |
Витаминотерапия |
• Террасная форма плеч |
|
|
Асимметричность атрофии! |
|
|
• Псевдогипертрофия икроножных и дельтовидных мышц |
|
|
• Умеренная выраженность контрактур и ретракций |
|
|
• Кардиомиопатия в редких случаях |
|
|
! Часто – аномалии сосудов сетчатки и снижение слуха |
|
|
|
|
|
129. Наследственные миотонии. Клиника, диагностика, лечение.
Миотонии: врождённая миотония, болезнь Лейдена-Томсена-Беккера и дистрофическая миотония Россолимо-Штейнерта-Куршмана
Болезнь Лейдена-Томсена-Беккера
1. Аутосомно-доминантный тип наследования – форма Томсена
-Дебют заболевания в возрасте 8-15 лет
-Первые признаки заболевания возникают в дистальных отделах рук, в последующем в процесс вовлекаются мышцы ног, жевательная и мимическая мускулатура
-«Атлетическое» телосложение
2. Аутосомно-рециссивный тип наследования – форма Беккера7
-Дебют заболевания в возрасте 4 -12 лет у девочек и ≈ 18 лет у мальчиков
-Первые признаки заболевания возникают в мышцах ног, через несколько лет - в мышцах рук + на поздних стадиях - лицевой мускулатуре (спазм круговых мышц).
*Миотонические феномены действия
1.замедленной релаксацией (длительной контрактурой) скелетных мышц после произвольного сокращения или электрической стимуляции
2.спазмы в различных группах мышц, возникающие после их интенсивного произвольного сокращения. Повторные движения облегчают расслабление, ослабляя спазм – «симптом врабатывания»
3.феномен усиливается на холоде, уменьшается - в тепле, во время отдыха и при приеме небольших доз алкоголя
*Перкуторные миотонические феномены
1.языка – после одиночного короткого удара молоточком формируются валик и ямка)
2.тенора – после одиночного короткого удара молоточком отведение и противопоставление большого пальца
БХ изменения при форме Томсена:
•При световой микроскопии – гипертрофия отдельных мышечных волокон.
•Уменьшение размеров II типа волокон.
•При тяжелых формах заболевания в мышечных волокнах обнаруживаются изменения в саркотубулярной системы, миофибриллярного аппарата, увеличение размеров и изменение формы митохондрий.
Дистрофическая миотония Россолимо-Штейнерта-Куршмана
Аутосомно-доминантный тип наследования
•Начало - 10-20 лет
•Сочетание симптомов миопатии, миотонии, сердечнососудистых, эндокринных нарушений и катаракты
•Спазмы преобладают в сгибателях пальцев кистей и жевательной мускулатуре. • Исчезновение на поздних стадиях
•Симптомы миопатии - мышцы лица, грудино-ключичнососцевидная мышца, надостные, подостные и височные мышцы
•Атрофические процессы максимально выражены в дистальных отдела конечностей.
•Бульбарные нарушения
•Атрофии симметричны
•У 50% больных - сердечно-сосудистые нарушения (нарушения проводимости, гипертрофия желудочков)
•Эндокринные нарушения - гипогонадизм, снижение либидо у мужчин и нарушение менструального цикла, гирсутизм и ранний климакс у женщин
•Характерно наличие алопеции (в области лба и висков у мужчин, гнездное или диффузное облысение у женщин)
•Характерно наличие катаракты, блефаритов, конъюнктивитов, помутнения роговицы. • Часто дизрафический статус
•У 30% больных интеллектуальная недостаточность
Диагностика – ЭНМГ ЭНМГ миотонические феномены:
1.Миотонические разряды
2.Единичные миотоничесие феномены
3.Звук «пикирующего бомбардировщика» Лечение:
• Мембраностабилизаторы: -хинидин -новокаинамид -дифенин
-карбамазепин (финлепсин) - мексилетин
• Ацетазоламид (диакарб)
• Препараты кальция (10 мл 10 % раствора хлорида кальция внутривенно, глюканат кальция внутримышечно
130. Спинальные амиотрофии. Симптомокомплекс «вялый ребенок» как ранний признак спинальных амиотрофий. Клиника, диагностика, лечение.
Патоморфология:
•Недоразвитие клеток передних рогов спинного мозга и двигательных ядер ствола
•Демиелинизация передних корешков и корешков ЧМН
•Пучковая атрофия скелетных мышц
Генетический дефект вызывает апоптоз клеток; утрата мотонейронов -> вялый паралич и денервационная атрофия поперечно-полосатых мышц Клиника общая:
•В большинстве случаев наблюдается симметричное поражение проксимальных мышц конечностей
•Дистальные амиотрофии, поражение бульбарной мускулатуры и асимметрия поражения развиваются реже
•Центральный мотонейрон обычно интактен
•Расстройств чувствительности не бывает
Острая злокачественная инфантильная спинальная амиотрофия 1го типа – форма Верднига-Гофмана
•Тип наследования – аутосомнорецессивный
•Дебют до 6 месяцев
•Генерализованная мышечная
•Гипотония, гипотрофия, гипоарефлексия, фасцикуляции
•Бульбарные расстройства
•Костно-суставные деформации, контрактуры суставов
•Врожденные пороки развития
•Быстро прогрессирующее течение
•Летальный исход к 2 годам
! ХАРАКТЕРАНЫ ПОЗЫ ЛЯГУШКИ И СИНДРОМ «ВЯЛОГО РЕБЁНКА» ! (Синдром вялого ребенка – врожденная мышечная гипотония)
Хроническая инфантильная спинальная амиотрофия 2го типа (промежуточная форма)
•Тип наследования – аутосомнорецессивный
•Начало в 6-24 месяца
•Подострое начало
•Быстропрогрессирующее заболевание
•Генерализованная мышечная гипотония, гипотрофия, гипоарефлексия, фасцикуляции
•Бульбарные расстройства
•Костно-суставные нарушения
•Летальный исход к 14-15 годам
Ювенильная спинальная амиотрофия 3го типа (форма Кугельберга-Веландера)
•Тип наследования – аутосомнорецессивный
•Дебют после 2 лет
•Постепенное начало
•Мышечная гипотония, гипотрофия, гипоарефлексия, фасцикуляции
•Псевдогипертрофии икроножных мышц
•Костные деформации (в 50%)
•Бульбарные нарушения не характерны
•Сухожильные ретракции и контрактуры в суставах редко
•Нарушение способности самостоятельной ходьбы через 10-12 лет
•Летальный исход в зрелом возрасте
Диагностика:
•Повышение уровня КФК (в 2-4 раза)
•Данные ЭНМГ (невральный (переднероговой) тип поражения)
•ДНК диагностика
ЭНМГ:
Для нейронального (переднерогового) типа поражения характерно:
-увеличение длительности и амплитуды ПДЕ;
-увеличение доли полифазных потенциалов;
-спонтанная активность регистрируется преимущественно в виде отдельных потенциалов фасцикуляции (ПФЦ)
Лечение:
1.ЛС-метаболики: Церебролизин, Цитофлавин, Глутаминовая кислота, АТФ, Карнитина хлорид, Метионин
2.Витамины группы В
3.Анаболические стероиды
4.ЛФК, масажи
5.Физиотерапии
6.Ортопедическая корррекция
131. Боковой амиотрофический склероз (БАС) и синдром БАС. Клиника, диагностика, лечение.
Наследуется по аутосомно-доминантному типу, иногда по аутосомно-рецессивному типу; встречаются спорадические случаи Дебют в возрасте 15-5- лет Патоморфология: дегенеративные изменения в передних рогах и боковых канатиках СМ + в бульбарном отделе ГС Начало процесса - шейно-грудной отдел
Клиническая картина:
1.переферические и центральные параличи + бульбарные расстройства
2.прогрессирующие атрофические параличи в дистальных отделах рук + формирование спастического пареза ног
3.фибриллярные подёргивания
4.поражение ядер XII XI X VII V пар ЧН
5.дегенерация обычно симметричная, но бывает и ассиметри симптомов
6.прогрессирует быстро
7.летальный исход вследствие сердечной/дыхательной недостаточности
Лечение - симптоматическое
1.Миорелаксанты и антиконвульсанты (снятие мышечной спастичности и болезненных судорог). Расслабляющие мускулатуру (баклофен, толперизон) и противосудорожные средства (карбамазепин, фенитоин).
2.Холиноблокаторы (при выраженном слюнотечении)
3.Декстрометорфан и хинидин - для коррекции бульбарных нарушений
4.Муколитики и отхаркивающие (при слабости дыхательной мускулатуры)
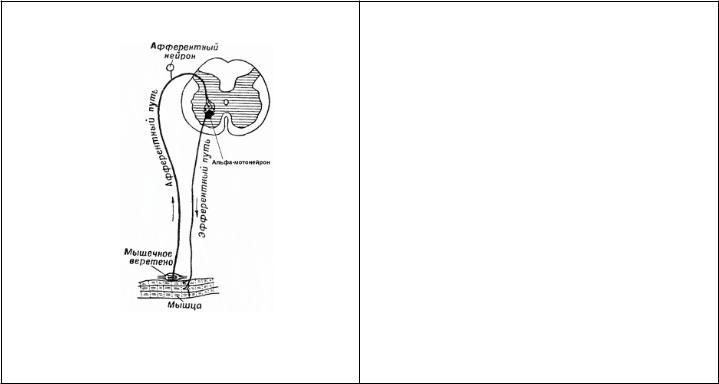
132.Наследственные невропатии (невральные амиотрофии). Клиника, диагностика, лечение.
Невральные амиотрофии - электрофизиологически и генетически гетерогенная группа заболеваний, объединенная клинической картиной полиневропатии
НЕВРАЛЬНЫЕ АМИОТРОФИИ ШАРКО-МАРИ-ТУТА (НМСН 1 И 2 ТИПА)
Патоморфология и клиника: 1 тип: дебют в 15-30 лет
Начало с дистальных отделов ног, симметрично, со временем вовлекаются руки
•Обнаруживается сегментарная демиелинизиция в периферических нервах
•Выявляются утолщения по типу «луковичных головок», (процессы ремиелинизации)
•В мышцах наблюдаются денервационные изменения с явлениями «пучковой» атрофии мышечных волокон 2 тип: позже Реже вовлекаются мускулатура рук, менее выражены чувствительные нарушения
Заболевание прогрессирует медленно, прогноз более благоприятный
•Обнаруживаются гибель аксонов периферических двигательных нервах и
•Вторичная сегментарная демиелинизиция
•Без формирования утолщений по типу «луковичных головок» (процессы ремиелинизации отсутствуют).
•В мышцах развиваются денервационные изменения с явлениями «пучковой» атрофии мышечных волокон.
Общая клиника:
Атрофии в мышцах голеней и стоп симметричны. «Симптом топтания».
Поражаются преимущественно перонеальная группа мышц и передняя большеберцовая мышцаформа ног «перевернутых бутылок» или «ног аиста».
Стопы деформируются, становятся «выеденными», «полая стопа». Походка больных - высоко поднимая ноги; ходьба на пятках невозможна. Кисти формы «когтистой лапы», «обезьяньей лапы».
+ Сенсорные и вегетативные нарушения.
Диагностика:
•Данные ЭНМГ (невральный тип поражения)
•ДНК диагностика
ЭНМГ: невральный тип поражения
-Увеличенние амплитуды ПДЕ;
-Увеличение длительности;
-Увеличение числа полифазных ПДЕ
-Спонтанная активность в виде положительных острых волн Лечение:
• Улучшение трофики мышц (АТФ, кокарбоксилаза, рибоксин, карнитина хлорид, церебролизин…)
• Улучшение проводимости по нервным волокнам (нейромидин)
• Витамины группы В, Е, А, липоевую кислоту
• Препараты, улучшающие микроциркуляцию (никошпан, пармидин, пентоксифиллин)
• ЛФК, массаж, физиотерапия
• Ортопедическое лечение
133. Наследственные нарушения метаболизма меди. Болезнь Менкеса. Болезнь Вильсона-Коновалова (гепатолентикулярная дегенерация). Клиника, диагностика, лечение. (Давыдов)
Болезнь Менкеса (болезнь курчавых волос)
Мутация гена, кодирующего Cu(2+)-транспортную АТФ-азу → нарушается клеточный транспорт меди Наследуется рецессивно, сцепленно с Х-хромосомой (проявляется только у мальчиков)
Клиника: Манифестирует в неонатальный период, наблюдается гипотермия. гипербилирубинемия, задержка физического развития, мб узелки на волосах, себорейный дерматит.
К 2-3 месяцам потеря приобретенных навыков, эпилептические приступы, спастический тетрапарез, патологические переломы рёбер, гиперрастяжимость кожи, диффузная гипопигментация кожи, гиперподвижность Диагностика: МРТ/КТ, микроскопия волос, снижение меди в сыворотке, при биопсии печени снижена медь, в слизистой кишечника повышено содержание меди Лечение: гистидинат меди 0,2-0,45 мг/день Прогноз неблагоприятный
Болезнь Вильсона-Коновалова
Аутосомно-рецессивный тип наследования. Происходит отложение меди в подкорковых узлах и в печени (+в коре БП, мозжечке, селезёнке, почках, роговице, хрусталике, радужке)
Причина- дефицит церулоплазмина, который участвует в транспорте меди Клиника: Дебют в 10-15 лет, нарастаем мышечная ригидность, разнообразные гиперкинезы, дрожание конечностей, головы,
туловища, дизартрия, изменения психики, иногда эпилептические припадки, увеличение и болезненность печени, признаки ПН. Специфический симптом: золотисто-зеленое или зеленовато-коричневое кольцо на радужке ( кольцо Кайзера-Флейшера) Диагностика: в крови снижен церулоплазмин, в моче дофига меди Лечение: препараты, связывающие медь (унитол, декаптол), снижающие мышечный тонус (циклодол, мидокалм), не кушать то, где много меди (орехи, печень, бобовые, орехи)
134. Наследственные болезни обмена веществ. Особенности клинических проявлений. Селективный биохимический скрининг и показания для его проведения. Подходы к терапии. (Давыдов)
Болезни обмена веществ- обширный класс моногенных заболеваний, обусловленных мутациями в генах, кодирующих ферменты, транспортные или сигнальные белки.
Суть: какое-то вещество накапливается (так как нет фермента для реакции) и становится токсичным, а то, в которое оно должно превратиться в недостатке и это приводит к нарушению функций клеток Особенности клинических проявлений:
-в половине случаев проявляется в раннем возрасте -задержка психического и двигательного развития -судороги -нарушения поведения (апатия или возбуждение)
-уменьшение показателей массы и роста -постепенная утрата приобретенных навыков
-прогрессивное нарастание неврологических нарушений -при многих заболеваниях сочетанное поражение НС, глаз, внутренних органов, ОПА -клиника часто неспецифична
-метаболические кризы→необратимые неврологические нарушения
Селективный биохимический скрининг
-это исследование, которое позволяет благодаря только одному анализу крови, взятому из пальца на специальную фильтровальную бумагу, диагностировать угрозу более 200 наследственных заболеваний из группы нарушений обмена веществ
Показания:
1.Внезапное ухудшение клинического состояния ребенка после периода нормального развития:
-острая метаболическая энцефалопатия
-летаргия (кома)
-судороги, резистентные к антиэпилептической терапии
2.Гепатомегалия
3.Метаболический ацидоз
4.Множественные переломы
5.Детская смертность в семье от заболеваний со сходными симптомами
6.Дополнительные критерии:
-кардиомиопатия
-гипогликемия
-тромбоцитопения
-повышение печеночных ферментов в 1,5 раза и более
-повышение КФК более чем в 2 раза выше нормы
-снижение ЩФ ниже возрастной нормы
-метаболический алкалоз
-повышение кетоновых тел в крови или в моче
-аномальный запах мочи/тела/ушной серы
-нарушение роста волос/алопеция
-резистентные к терапии судороги
-костно-суставные аномалии (тугоподвижность суставов, деформация грудной клетки)
-грыжи
-частые срыгивания/рвота
-дистонии/гиперкинезы
Заболевания, выявляемые при селективном скрининге:
1.Фенилкетонурия
2.Гомоцистинурия
3.Тирозинемия, тип 1
4.Болезнь кленового сиропа
5.Глутаровая ацидурия
6.Пропионовая ацидурия
7.Метилмалоновая ацидурия
8.Изовалериановая ацидурия
9.Недостаточность биотинидазы
135.Наследственные болезни обмена веществ. Болезнь Фабри. Клиника, диагностика, лечение.
Ферментозаместительная терапия. (Давыдов)
Болезнь Фабри
Сфинголипидоз, наследственное нарушение метаболизма, вызванное дефицитом альфа-галактозидазы А. Группа лизосомных болезней накопления.
Наследуется: Х-сцепленно Эпидемиология: 1:500 000 человек
Патогенез: нарушается накопление гликофосфолипидов в лизосомах клеток (эндотелий, клетки сердца, почек, нервной системы).
Характер: мультисистемный прогрессирующий Дебют: 2-4 года/подростковый возраст Клинические проявления:
1.Ангиокератомы
2.Полиневропатии
3.Хроническая, изнуряющая, жгучая боль в конечностях
4.Гиперпатия – чрезмерная болевая реакция на болевой стимул
5.Акропарестезия
6.Снижение или полное отсутствие потоотделения
7.ТИА уже в малом возрасте (геморрагический инсульт, ишемические инсульты)
8.Повреждение почек и хроническая почечная недостаточность
9.Поражение сердца (ГЛЖ)
10.Помутнение роговицы («мутовчатое» помутнение роговицы)
Диагностика:
1.Активность АГАЛ в пятнах крови
2.Молекулярно-генетическое исследование
3.Концентрация глоботриаозилсфингозина в крови
4.МРТ
Лечение:
Ферментозаместительная терапия (ФЗТ) препратами агалсидаза альфа, агалсидаза бета.
Цель терапии: а) уменьшение невропатической боли; б) регресс гипертрофии ЛЖ; в) стабилизация функции почек