

nevra_esche_ekzamen
.pdfРис. 4.6. Мигрирующие нейроны:
а — в — последовательные этапы миграции
В самой нервной трубке также происходят изменения. В конце третьей недели развития ее ростральный конец преобразуется в мешковидное расширение, дающее начало ГМ, а каудальный отдел дает начало СМ. На головном конце нервной трубки формируются три расширения — три первичных мозговых пузыря (стадия трех мозговых пузырей). Полости этих пузырей, несколько изменяя форму, сохраняются во взрослом мозгу в виде мозговых желудочков и мозгового водопровода. Самым ростральным пузырем является prosencephalon, или первичный передний мозг, за ним следует mesencephalon — первичный средний мозг, последний пузырь, за которым уже начинается СМ, это rhombencephalon — первичный задний мозг (рис. 4.6).
Рис. 4.6. Стадии трех (а) и пяти (б) мозговых пузырей
На втором месяце развития первый и третий первичные мозговые пузыри с помощью борозд разделяются, образуя каждый по два вторичных мозговых пузыря (стадия пяти мозговых пузырей). Prosencephalon делится на telencephalon — конечный мозг (полушария большого мозга и базальные ядра) и diencephalon — промежуточный мозг. С каждой стороны промежуточного мозга вырастает глазной пузырь, формирующий нервные элементы сетчатки глаза. Глазной бокал, образованный этим выростом, вызывает изменения в лежащей непосредственно над ним эктодерме, что приводит к отделению от нее клеток, образующих хрусталик. Rhombencephalon разделяется на metencephalon — собственно задний мозг, включающий мозжечок и варолиев мост, и myelencephalon — продолговатый мозг. Средний мозг сохраняется как единое целое.
Отдельные части нервной трубки растут с разной скоростью. В результате этого одновременно с формированием пяти мозговых пузырей образуются изгибы зачатка головного мозга: сначала — среднемозговой (основной) изгиб в области среднего мозгового пузыря, обращенный выпуклостью дорсально; потом шейный изгиб на границе головного и спинного
мозга, также выпуклостью дорсально; последний — мостовой изгиб в области заднего мозгового пузыря выпуклостью вентрально (рис. 4.7).
После формирования мозговых пузырей в структурах ЦНС происходят сложные процессы внутренней дифференцировки и роста.
Г 
Рис. 4.7. Изгибы зачатка головного мозга (пятая неделя развития)
Ввозрасте 10—20 недель образуются все основные отделы НС. К этому моменту заканчивается миграционный период ее развития, т.е. все нейроны перемещаются туда, где они будут находиться во взрослом мозгу. Полушария постепенно становятся самой большой частью НС, происходит выделение основных долей (образование борозд и извилин происходит во второй половине эмбриогенеза). Из оболочек в ткань мозга врастают кровеносные сосуды. В спинном мозгу формируются шейное и поясничное утолщения. Окончательный вид приобретает мозжечок.
Впоследние месяцы эмбрионального развития в НС заканчивается формирование внутренней структуры мозга (его ядер и трактов). Активно идут процессы синаптогенеза (образования синапсов), благодаря чему формируются рефлекторные дуги многих безусловных рефлексов. Начинается активная миелинизация сначала спинного (в возрасте 20 недель), а затем (в возрасте 36—40 недель) головного мозга, которая в основном заканчивается только к 10—12 годам. Отметим, что миелинизация начинается позднее в филогенетически более молодых структурах. Кора больших полушарий к моменту рождения более или менее развита и уже обладает характерной складчатой поверхностью.
Надо отметить, что изначально в НС образуется избыточное количество нейронов. Окончательное число нейронов определяется запрограммированной гибелью клеток как до, так и после рождения. В разных областях мозга количество погибших нейронов может колебаться от 15 до 85%. По имеющимся данным, гибель связана с конкуренцией между нейронами, и их выживание прямо зависит от функциональной активности каждой конкретной клетки.
Мозг новорожденного весит примерно 350 г, т.е. 10% всей массы тела. Вес мозга взрослого человека равен в среднем 1300 г (2% массы тела). Так как деление большинства нервных клеток прекращается еще до рождения, увеличение массы мозга происходит за счет роста тел нейронов и их отростков, образования новых синапсов, миелинизации нервных волокон, деления и роста клеток нейроглии. Мозг растет главным образом в течение первого года жизни, когда его вес увеличивается примерно до 1000 г. Очень показательно, что синаптогенез наиболее активно идет именно в первые годы жизни. Дендритное дерево у двухлетнего ребенка гораздо больше отличается от новорожденного, чем от взрослого (рис. 4.8).

Рис. 4.8. Развитие дендритного дерева после рождения
После 50—60 лет начинаются структурные и химические изменения мозга. Общее число нейронов снижается, но в разных областях мозга этот процесс проходит неравномерно. Например, в гипоталамусе, который регулирует жизненно важные функции, исчезает очень мало нейронов. Даже когда нейроны сохраняются жизнеспособными, их тела и отростки могут атрофироваться. Чаще это происходит в структурах мозга, участвующих в сложных психических процессах (запоминании, обучении, планировании действий). Однако, по-видимому, мозг обладает значительным физиологическим резервом, позволяющим компенсировать потери и повреждения нейронов. Показано, что мозг 80-летних здоровых людей почти также активен, как и мозг 30-летних.

105. Ген, свойства гена как единицы функционирования. Генотип человека. Генетический груз у человека. Основные признаки наследственной патологии. Аппакова
Ген – элементарная единица наследственности, наименьший неделимый элемент наследственного материала, который может быть передан от родителей потомству как целое и который определяет признаки, свойства или физиологическую функцию организма.
●Участок ДНК, на котором закодирована информ о синтезе опред белка
Свойства:
●Дискретность действия (каждый ген действ как самостоятельная единица наследств)
●Стабильность (нет мутаций => передается в ряду поколений без изменений)
●Специфичность действия (каждый ген = свой признак)
●Плейотропность (один ген может обеспечивать развитие неск признаков)
●Лабильность (способность к мутациям)
●Экспрессивность (степень выраженности признака)
●Пенетрантность (частота проявления гена среди его носителей)
Генотип – это полный набор генетического материала организма. В более узком смысле под генотипом понимают комбинацию аллелей гена или локуса у конкретного организма. Генотип вместе с факторами внешней среды определяет фенотип организма.
●генотип особей каждого вида является целостной системой, хотя состоит из отдельных генов, которые могут отделяться друг от друга и наследоваться независимо.
Генетический груз = патологические генные мутации, наследуемые от родителей и прародителей, и называемые сегрегационным грузом, если в виде болезни проявляются рецессивные или нелетальные доминантные мутации генов (ИЛИ
накопление летальных и сублетальных отрицательных мутаций, вызывающих при переходе в гомозиготное состояние выраженное снижение жизнеспособности особей, или их гибель)
●Генетический груз рассматривается как мера неприспособленности популяции к условиям окружающей среды
●Пример: аллели мутантных форм Hb = HbC, HbS
Основные признаки наследственной патологии.
●Семейный хар-р заболевания
●Хроническое, прогрессирующее, рецидивирующее течение
●Специфические с-мы наследств болезней
●Патологические изменения органов и систем (системность заболевания)
●Врожденный хар-р заболевания
●"Резистентность к наиболее распр методам лечения
106. Нормальное число хромосом в хромосомном наборе у человека. Структурная организация хромосомы. Цитогенетические и молекулярногенетические методы исследования. Показания к проведению.
Кариотип человека - совокупность морфологических особенностей полного хромосомного набора ядра соматической клетки, свойственная виду Homo sapiens (для женского пола - 46,ХХ; для мужского пола - 46,XY, т.е. двойной, диплоидный набор хромосом, включающий 44 аутосомы и 2 половые хромосомы - гоносомы).
Хромосомы – морфологические единицы ядра эукариотической клетки, функции которых состоят в хранении, реализации и передаче наследственной информации.
Компактизация ДНК: Двойная спираль ДНК вследствие образования комплекса с гистонами образует нуклеосомную нить, которая затем скручивается в спираль - хроматиновую фибриллу. Укладка хроматиновой фибриллы в петли с участием негистоновых белков образует интерфазную петельную хроматиновую фибриллу. Дальнейшая компактизация приводит к образованию хроматид. Метафазная хромосома состоит из 2 хроматид (2 молекулы ДНК), которые в анафазе митоза расходятся к разным полюсам и попадают в дочерние клетки. Длина молекулы ДНК в одной хромосоме человека приблизительно равно 4см. Длина метафазной хромосомы около 6 мкм
Строение метафазных хромосом: функциональными элементами хромосомы эукариот являются центромера, теломеры, сайты инициации репликации.
1.В области центромеры сестринские хроматиды (дочерние молекулы ДНК) прикрепляются к митотическому веретену деления, что обеспечивает их точное расхождение в дочерние клетки в митозе.
2.На центромере происходит сборка кинетохора – сложной белковой структуры, определяющей прикрепление хромосомы к микротрубочкам веретена деления., обеспечивающим движение дочерних хромосом в митозе.
3.Центромера делит хромосомы на две части: короткое плечо (p) и длинное плечо (q).
4.В зависимости от расположения центромеры хромосомы могут быть метацентрическими, субметацентрическими, акроцентрическими и телоцентрическими.

Некоторые хромосомы на конце плеча имеют вторичную перетяжку (ядрышковой организатор – на рисунке – «ножка»), которая содержит рибосомные гены и отделяет от хромосомы «спутник». Ядрышковые организаторы есть у акроцентрических хромосом 12, 14, 15, 21, 22.
Методы исследования:
К признакам, характеризующим кариотип относят число, размер, форму хромосом, положение центромеры, наличие вторичных перетяжек, чередование гетеро- и эухроматиновых участков.
1.Изучение кариотипа проводится с применением цитогенетического метода. Используют соматические клетки,
имеющие ядро и способные делиться. Клетки помещают в культуру, стимулируют их деление. Блокированные в метафазе клетки фиксируют и окрашивают. Существуют различные методики окраски хромосом: рутинная, дифференциальная, FISH-метод и др. Результаты идентификации хромосом на препарате представляют в виде идиограммы. Использование дифференциальной окраски хромосом позволило разделить q и p плечи хромосом, а в них выделить сегменты(Парижская классификация хромосом).
Цитогенетический метод позволяет обнаружить аномалии числа хромосом и наличие структурных перестроек.
 - идиограмма
- идиограмма
2.Молекулярно-генетические методы направлены на выявление изменений в определенных участках ДНК генов или хромосом.
●Базовые методы : в основе многочисленных методов ДНК-диагностики лежат два базовых метода:
блот-гибридизационный анализ и ПЦР.
Блот-гибридизационный анализ - это самый первый метод исследования молекулы ДНК.
1.ДНК, выделенная из биоптата органа, ткани или культуры лимфоцитов периферической крови, подвергается расщеплению рестриктазами, которые «узнают» в ней строго определенную последовательность нуклеотидов.
2.Далее из множества нарезанных фрагментов нужно найти один или несколько фрагментов ДНК с определенной последовательностью и охарактеризовать их.
3.Затем разделенные фрагменты «перепечатывают» на целлюлозный фильтр или нейлоновую мембрану, фиксируют и подвергают гибридизации с зондом (олигонуклеотидом), интенсивно меченным радиоактивным изотопом или флуоресцентной меткой.
4.Далее следует отмывка несвязанной с фильтром радиоактивной метки и экспозиция фрагмента на рентгеновской пленке.
ПЦР идет в четыре этапа на специальных программных термоциклерах, задающих и поддерживающих определенный температурный режим реакции.
Компонентами этой реакции служат: матричная ДНК, олигопраймеры, смесь дезоксинуклеотидов и термофильная ДНКполимераза. Их добавляют в специальный солевой буфер непосредственно перед помещением пробирки в термоциклер.
1.На первом этапе реакции двухнитевая матричная ДНК переводится в однонитевую форму при нагревании в течение нескольких минут до температуры 95-98 °С.
2.На втором этапе (этап гибридизации) температура реакционной смеси снижается до 30-50 °С, и олигопраймеры гибридизируются с денатурированной (одноцепочечной) ДНК, содержащей комплементарные участки.
3.На третьем этапе температура повышается до 60-70 °С (она оптимальна для термофильной ДНК-полимеразы), что позволяет запустить синтез ДНК в направлении от 5'- к 3'-концу геномной ДНК-матрицы.
4.На четвертом этапе происходит дальнейшее повышение температуры до 80-90 °С, что прекращает синтез ДНК, и начинается денатурация с освобождением синтезированных фрагментов ДНК с геномной матрицы.
5.В результате образовавшиеся фрагменты ДНК становятся матрицами для следующих циклов амплификации, и
процесс может протекать в геометрической прогрессии, давая необходимое количество циклов.
Анализ результатов ПЦР проводится с помощью гельэлектрофореза продуктов амплификации, которые в случае необходимости обрабатываются рестриктазами. В дальнейшем гели окрашивают бромидом этидия и исследуют продукты рестрикции в проходящем УФ-свете с длиной волны 380 нм на наличие в них невыявленных мутаций.
Показания к проведению:
1.Нарушения репродуктивной функции неясной этиологии у мужчин и женщин:
●спонтанные аборты (два и более) на разных сроках;
●мертворождения или рано умершие дети;
●первичная аменорея.
2.Мужское и женское бесплодие при исключении гинекологических заболеваний у женщин
3.Множественные врожденные пороки развития у ребенка или задержка развития ребенка
4.Подозрение на хромосомную болезнь на основании клинических симптомов
5.Пренатальная и предимплантационная диагностика при повышенном генетическом риске
6.Подозрение на синдромы, характеризующиеся хромосомной нестабильностью (учет хромосомных аберраций и сестринских хроматидных обменов)
7.Гемобластозы и онкологические заболевания
8.Оценка мутагенных воздействий
107.Фенотип нормальный и патологический. Наследственная и врожденная болезнь. Генетическая и негенетическая
болезнь. Методы их диагностики.
Фенотип - это совокупность всех признаков организма, обусловленных совместным действием генотипа и факторов среды.
Нормальный фенотип - это совокупность всех нормальных признаков организма, обусловленных нормальным действием генотипа и факторов среды (результат их взаимодействия).
Патологический фенотип - это наличие ряда патологических признаков организма, обусловленных патологическим действием генотипа и факторов среды (результат их взаимодействия), на фоне других нормальных признаков организма.
Здесь следует пояснить смысл формулировки «...на фоне других нормальных признаков»:
Если у больного человека выявлен конкретный патологический признак или фенотип (например, симптоматика ОРВИ), то это совсем не означает, что у него исчезли другие (нормальные) признаки, например голубая окраска глаз, курчавость волос и др.
Патологический фенотип как симптомокомплекс болезни - это результат совместного патологического действия генотипа и факторов окружающей среды.
Фенотипический полиморфизм - это многообразие нормальных и патологических признаков и фенотипов, выявляемых на любых уровнях дискретности организма: молекулярном, клеточном, тканевом, органном и организменном.
Наследственная болезнь - это возникший в ходе онтогенеза постоянный (конституциональный) патологический фенотип с признаками патокинеза и прогредиентности, передаваемый из поколения в поколение.
Врожденная болезнь - это возникший внутриутробно постоянный патологический фенотип без признаков патокинеза и прогредиентности, передаваемый или не передаваемый из поколения в поколение, что связано с генетической или негенетической причиной болезни. Например, если диагноз синдрома Дауна поставлен уже при рождении ребенка, то фенотип такого больного остается стабильным в течение всей его жизни, ибо он обусловлен хромосомным нарушением.
Методика обследования больных с наследственной патологией включает: первый (клинический) и второй (параклинический) этапы.
На клиническом этапе применяются: основные и дополнительные клинические, а также основные клинико-инструментальные и клинико-лабораторные методы. Кроме того, на завершающей стадии этого этапа применяется метод постановки предварительного диагноза на основе компьютерных баз данных. В случае обследования организма беременной, зародыша, эмбриона и плода дополнительно применяются специальные клинико-инструментальные и клинико-лабораторные методы преимплантационной и пренатальной диагностики.
На параклиническом этапе применяются специальные лабораторные методы, цитогенетические, молекулярно-генетические и биохимические методы, а в определенных ситуациях еще и популяционно-статистический метод
МЕТОДЫ ПЕРВОГО ЭТАПА ДИАГНОСТИКИ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ
Основные клинические методы
●первичный прием у врача (обязателен сбор данных о сибсах, родственников 1 и 2 поколений как минимум, предыдущих беременностях, родах, периодах новорожденности, раннего развития, наличие тератогенных и мутагенных факторов в процессе беременности матери, профессиональные вредности родителей, ИППП, соматические болезни)
●клинико-генеалогический метод (сбор врачом генеалогических данных, необходимых для последующего составления и анализа семейной родословной пробанд).
●объективный врачебный осмотр (основой такого осмотра является семиотика - наука о знаках (символах) патологического процесса в организме. Во многих случаях у больного с наследственной патологией наблюдается характерный внешний вид, напр кошачий крик, глаза лани, лицо эльфа)
●синдромологический метод – «поэтажное» обследование пробанда с выявлением признаков наследственной патологии. Патогномоничные, ведущие и типичные признаки.
●Далее врач использует основные клинико-инструментальные и клинико-лабораторные методы.
●Дополнительные методы: близнецовыйсравнение монозиготных и дизиготных близнецов; дерматоглифика. В настоящее время эти методы применяются редко.
Первый этап диагностики завершается постановкой предварительного диагноза. Используются мировые базы данных наследственных патологий. Напр, каталог менделирующих признаков человека В. Мак-Кьюсика (OMIM); Лондонская (Оксфордская) медицинская база данных (LMD).
К специальным клинико-инструментальным и клинико-лабораторным методам относятся методы
преимплантационной( с помощью микрохирургического вмешательства от зародыша отделяются одна или две клетки для последующего генетического анализа) пренатальной диагностики ( УЗИ, ЭХОКГ, фетоскопия, фетрамниография, амниоцентез, биопсия ворсин хориона, кордоцентез) наследственных и врожденных болезней.
Перед вторым этапом обследования больных с наследственной патологией врач может принять решение о применении популяционно-статистического метода, который не относится ни к методам первого этапа, ни к методам второго этапа диагностики наследственной патологии.
●Популяционно-статистический метод - это общий эпидемиологический метод, связанный с изучением частоты распространения наследственной патологии и проявлений ее полиморфизма в разных популяционных группах.
МЕТОДЫ ВТОРОГО ЭТАПА ДИАГНОСТИКИ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ
Второй этап диагностики наследственной патологии связан с использованием параклинических методов.
●методы аналитической биохимии
1.качественные - с их помощью определяют избыточную концентрацию субстратов заблокированных ферментных реакций накапливающихся при наследственных болезнях обмена. Например, органолептический тест на специфический цвет и запах при алкаптонурии, 3-гидроксил-3-метилглутаровой ацидурии, изовалериановой ацидемии, лейцинозе, тирозинемии, ФКУ, цистинурии.
2.Количественные - С их помощью определяют изменения концентрации веществ и нарушения кислотно-щелочного баланса. Применяются и для массовой и селективной диагностики НБО у новорожденных. Напр, ЦПХ-тест при мукополисахаридозах.
●иммунологические методы: анализ гамма-глобулинов, В- и Т- лимфоцитов, содержания комплемента в крови, исследование функции фагоцитарных клеток, определение HLA-антигенов в лейкоцитах и др.
●цитогенетический метод см. пред. вопрос + применяются для кариотипирования и цитогенетического анализа хромосомного набора пробанда, его больных и здоровых родственников
●молекулярно-генетические методы обследования пробанда, его больных и здоровых родственников: относятся к основным диагностическим тестам, применяемым в клинической генетике последних лет. См. основную информацию в предыдущем вопросе + следующее: Амплификации рефрактерной мутационной системы, ПЦР-опосредованный сайт-направленный мутагенез, Лигирование синтетических однонуклеотидных зондов, Анализ конформационного полиморфизма однонитевой ДНК или выявление точковых мутаций, Денатурирующий градиентный гель-электрофорез, Гетеродуплексный анализ, Химическое расщепление некомплементарных сайтов, Денатурирующая жидкостная хроматография высокого разрешения
108. Варианты наследования генов и признаков: моногенное, полигенное, неклассическое («материнское» наследование, импринтинг, экспансия числа нуклеотидных повторов).
Моногенное наследование основано на первом и втором законах наследственности. Оно подразумевает наследование одного гена (одной пары признаков) и относится к аллельным генам.
Типы моногенного наследования:
●аутосомно-доминантный тип
●аутосомно-рецессивный тип
●Х-сцепленный доминантный тип, напр. витамин D-резистентный рахит
●Х-сцепленный рецессивный тип, напр. гемофилия А и В
●Y-сцепленный тип или голандрическое наследование, напр. нарушения дифференцировки пола
Полигенное наследование основано на третьем законе наследственности. Оно подразумевает наследование двух генов (пар признаков) и более и относится к неаллельным генам.
●Полигенное наследование нередко называют мультигенным или мультифакториальным, имея в виду наследование одновременно не одного, а нескольких определенных генов, проявляющих свое действие в специфических условиях окружающей среды, при наличии провоцирующих факторов.
Критерии полигенного наследования:
1.Наследуемость признака или болезни. Чем выше наследуемость признака или заболевания (чем больше унаследовано генов, за него ответственных), тем выше риск его развития у здоровых родственников.
2.Степень выраженности признака или тяжесть течения болезни.
3.Общность генов у пробанда и его родственников (или близкая степень родства с больным родственником)
4.Редко поражаемый пол. Мультифакториальный признак или заболевание проявляется чаще у лиц редко поражаемого пола (критерий, названный эффектом Картера).
5.Число больных родственников
Нетрадиционное наследование – наследование генов и признаков, выходящее за рамки моногенного и полигенного вариантов.
Плохо изученные или вообще неизвестные заболевания 6 классов:
1.болезни накопления
2.пероксисомные болезни
3.митохондриальные болезни
4.болезни импринтинга
5.болезни экспансии числа нуклеотидных повторов
6.прионные болезни.
При наследовании заболеваний первых трех классов речь идет о материнском наследовании, т.к. лизосомы, митохондрии и пероксисомы, являясь цитоплазматическими структурами соматических клеток, наследуются исключительно по линии мать-дочь и никогда не передаются по линиям мать-сын, отец-сын или отец-дочь, что обусловлено биологическим матриархатом.
В случае болезней импринтинга или эпигеномного выключения из экспрессии локусов хромосом одного из родителей фенотипические проявления действия гена могут меняться в результате трех причин:
1.делеция гена (генетический импринтинг)
2.однородительская изодисомия (хромосомный импринтинг)
3.нарушение генной экспрессии в центре импринтинга
Импринтинг – результат качественных, а не количественных изменений наследственного материала.
1.Генетический импринтинг основан на механизме специфического метилирования цитозиновых оснований молекулы ДНК, выключающем транскрипцию гена.
2.Геномный импринтинг основан на однородительской изодисомии по конкретной хромосоме либо материнского, либо отцовского происхождения. Напр, синдром Ангельмана.
3.Импринтинг в результате ошибок генной экспрессии в центре импринтинга происходит также при синдромах Ангельмана и Прадера-Вилли. Он обусловлен тем, что оба родителя передают больному потомку гены, несущие их специфические свойства, т.е. гены отца и матери активированы или супрессированы у потомка по-разному (так называемые импринтированные гены).
Экспансия нуклеотидных повторов – патологическое увеличение числа копий коротких нуклеотидных последовательностей ДНК, которые обычно связаны с тяжелыми неврологическими заболеваниями. Эти типы мутаций нестабильны и динамичны, а количество повторов может меняться от поколения к поколению. Процесс экспансии копий нуклеотидных повторов связан с нарушениями систем репарации ДНК
Болезни экспансии нуклеотидных повторов, связанных с увеличением числа кодирующих и некодирующих последовательностей нуклеотидов в результате динамических мутаций. Динамическая мутация «движется» от состояния фенотипически не проявляющейся премутации к состоянию фенотипически проявляющейся полной мутации. Такие мутации лежат в основе ряда тяжелых наследственных нейродегенеративных заболеваний. Например, повтор ЦЦГ характерен для синдрома Мартина-Белл (Xq23), повтор ЦТГ - для спинобульбарной мышечной атрофии (Xq11-12); ЦАГ - для миотонической дистрофии (19q13.3); ЦТГ - для хореи Гентингтона (4р16.3).
109. Мутационный процесс и мутации. Классификация мутаций.
Мутационный процесс или мутагенез, есть непрерывно идущий процесс формирования мутаций под действием мутагенов - факторов среды, повреждающих наследственный материал.
Впервые теория непрерывно идущего мутагенеза предложена в 1889 г. русским ученым из Петербургского университета С.И. Коржинским.
●спонтанный мутагенез: мутации происходят без видимых внешних причин, но под влиянием внутренних условий в клетке и организме.
●индуцированный мутагенез: вызванные искусственно путем воздействия внешних факторов физической,
химической или биологической природы.
Вновь возникшие мутации называются новыми мутациями или мутациями de novo.
Мутация - изменение структурной организации, количества и/или функционирования наследственного материала и синтезируемых им белков. Это понятие впервые предложил Гуго де Фриз, он же описал свойства мутаций:
●возникают внезапно;
●передаются из поколения в поколение;
●наследуются по доминантному типу (проявляются у гетерозигот и гомозигот) и рецессивному типу (проявляются у гомозигот);
●не имеют направленности («мутирует» любой локус, вызывая незначительные изменения или затрагивая жизненно важные признаки);
●по фенотипическому проявлению бывают вредными (большинство мутаций), полезными (крайне редко) или безразличными;
●возникают в соматических и половых клетках.
Кроме того, одни и те же мутации могут возникнуть повторно.
Прямые мутации: переход от нормального состояния гена (признака) к патологическому состоянию, реверсия – наоборот.
Последующие мутации в гене, подавляющие первичный мутантный фенотип, называются супрессорными.
Мутации: соматические и герминативные. Недавно было доказано, что соматические мутации могут наследоваться. Напр, мультифакториальные формы рака.
Мутагенез: радиационный, химический, биологический.
Классификация мутаций
Взависимости от уровня организации наследственного материала:
●Точковые: изменения нуклеотидов (оснований) одного гена, ведущие к изменению количества и качества синтезируемых ими белковых продуктов. Изменения оснований - это их замены, вставки, перемещения или выпадения, которые можно объяснить мутациями в регуляторных областях генов (промотор, сайт полиаденилирования), а также в кодирующих и некодирующих областях генов (экзоны и интроны, сайты сплайсинга).
К ним относят мутации транскрипции(первого типа) и мутации трансляции (второго типа).
●Структурные: нарушают структуру хромосом и приводят к формированию новых групп сцепления генов. Это делеции (утраты), дупликации (удвоения), транслокации (перемещения), инверсии (поворот на 180°) или инсерции (вставки) наследственного материала. Такие мутации характерны для соматических клеток, включая стволовые клетки. Наиболее известные примеры: синдром «кошачьего крика» (кариотип: 46,ХХ,5р-), синдром ВольфаХиршхорна (46,ХХ, 4р-), транслокационная форма синдрома Дауна (кариотип: 47, ХУ, t (14;21)).
●Геномные мутации - нарушение числа хромосом или их частей (ведут к появлению новых геномов или их частей путем добавления или утраты целых хромосом или их частей). Происхождение этих мутаций обусловлено нерасхождением хромосом в митозе или мейозе.
Впервом случае (митоз) - это анеуплоиды, тетраплоиды с неразделенной цитоплазмой, полиплоиды, имеющие по 6, 8, 10 пар хромосом и более. Во втором случае - это неразделение парных хромосом, участвующих в формировании гамет (моносомии, трисомии) или оплодотворение одной яйцеклетки двумя сперматозоидами (диспермия или триплоидный зародыш). Их примерыэто синдром Шерешевского-Тернера (45,ХО), синдром Клайнфельтера (47,ХХУ), регулярная трисомия при синдроме Дауна (47,ХХ, +21).
Классификация мутаций в зависимости от их фенотипического эффекта.
1.аморфные мутации. Это состояние, при котором признак, контролируемый патологическим аллелем, не проявляется, так как патологический аллель не активен по сравнению с нормальным аллелем. Напр, альбинизм.
2.антиморфные мутации. В этом случае значение признака, контролируемого патологическим аллелем, противоположно значению признака, контролируемого нормальным аллелем.
3.гиперморфные мутации. В случае такой мутации признак, контролируемый патологическим аллелем, выражен сильнее признака, контролируемого нормальным аллелем.
4.Гипоморфные мутации. Это состояние, при котором проявление признака, контролируемого патологическим аллелем, ослаблено по сравнению с признаком, контролируемым нормальным аллелем. Напр, анемия Фанкони.
5.Неоморфные мутации. О такой мутации говорят, когда признак, контролируемый патологическим аллелем, будет иного (нового) качества по сравнению с признаком, контролируемым нормальным аллелем. Пример: синтез новых иммуноглобулинов в ответ на проникновение в организм чужеродных веществ.
110. Клинический этап диагностики наследственной патологии. Особенности осмотра. Клинико-генеалогический метод. Синдромологический метод.
Основные клинические методы Первичный прием у врача – предусматривает работу с пробандом (консультируемое лицо) и его ближайшими
родственниками. Во время первичного приема проводятся сбор данных анамнеза жизни и анамнеза заболевания и их анализ.
Вчисле данных должны быть собраны сведения о:
●сибсах (братья и сестры), родителях и других родственниках пробанда, как минимум I и II степеней родства;
●предыдущих беременностях и родах, периодах новорожденности, вскармливания и раннего развития пробанда;
●отягощенном характере течения данной беременности (прием лекарств, воздействие мутагенных и тератогенных факторов на эмбрион и плод, наличие у супругов вредных привычек и профессиональных вредностей);
●отягощенном акушерском анамнезе (наличие токсикозов, угрозы выкидыша, многоили маловодия, преждевременных родов, случаев предшествующего мертворождения или рождения детей с ЗВУР, ВПР, ХБ, МБ и МФЗ)
●отягощенном гинекологическом анамнезе (инфекции, передаваемые половым путем, хронические соматические заболевания неинфекционной природы).
Кроме данных о пробанде, врач собирает и анализирует данные о его близких родственниках, имеющих подобное заболевание или частично сходную симптоматику. Если такие родственники выявляются, то они приглашаются для объективного обследования.
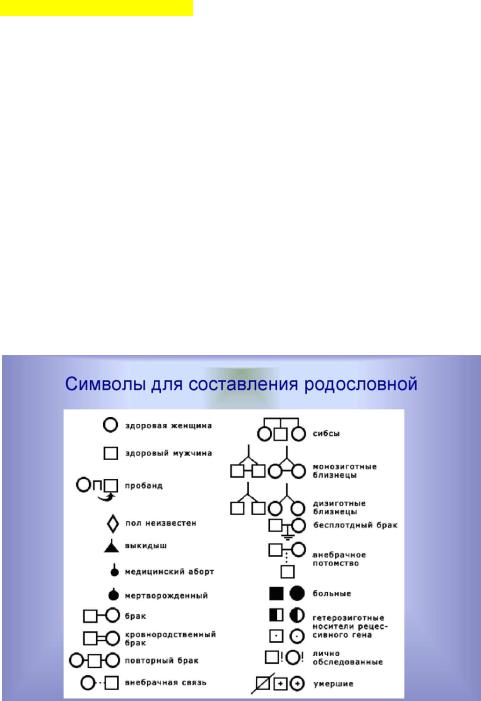
Клинико-генеалогический метод
Сбор врачом генеалогических данных, необходимых для последующего составления и анализа семейной родословной пробанда. Сбор таких данных нацелен на выявление и изучение симптомов наследственного и врожденного заболевания, проявившихся у пробанда, а также у его больных и здоровых родственников. В ходе сбора данных желательно использование семейных фотоальбомов, медицинских архивов и другой документации.
При составлении родословной врач применяет стандартные приемы и символы
.