

Полезные материалы за все 6 курсов / Учебники, методички, pdf / Шапошников_Травматология_и_ортопедия_3_том
.pdfА— Д, в третьих X — Р или X — Д (псевдоахондроплазия — А — Д,
А— Р; множественная эпифизарная дисплазия — А — Д, А — Р; спондилоэпифизарная дисплазия — А — Д,А — Р, X — Р). Причина заключается в том, что любая особенность организма является резуль татом осуществления длинной цепи реакций, каждая из которых уп равляется особым геном, и что блокада (дефект) любого звена данной цепи реакции в конечном итоге приведет к той же аномалии. Следо вательно, при разных типах наследования клинически однородного заболевания внешне сходная клиническая картина может быть связана с мутацией разных генов.
Основной молекулярно-генетический дефект для большинства НЗС неизвестен. Изучаются наследственно обусловленные количе ственные или качественные клеточные изменения, а также нару шения процессов биосинтеза и распада компонентов межклеточного матрикса соединительной ткани (коллагеновых и эластических во локон, протеогликановых комплексов), которые проявляются в из менениях структуры и функции соединительной ткани и ее специ ализированных типов (кожа, кость, хрящ). Тактика поиска моле кулярного дефекта исходит из того, что причинами возникновения НЗС могут быть структурные и регуляторные мутации генов (пер вичные причины); изменения метаболизма в клетках и во внекле точном пространстве, обусловленные различными энзимопатиями (вторичные причины); нарушение фенотипической экспрессии кле ток, синтезирующих коллаген и/или другие компоненты межкле точного матрикса. В отношении остеохондродисплазий (ОХД) наи большие успехи достигнуты в исследовании генетико-биохимических дефектов при НО. Они связаны с изменениями в первичной струк туре коллагена преимущественно типа I, вызываемыми мутациями
вгенах, кодирующих синтез проколлагеновых цепей типа I, и приводящими к нарушению процесса фибриллогенеза. Локализация генов, контролирующих синтез двух проколлагеновых цепей кол лагена типа I, в настоящее время установлена: хромосомы 17 и 7. Однако не исключено, что клинический полиморфизм НО обуслов лен различным уровнем локализации причины дефекта.
Блокада синтеза, процессинга или деградации протеогликанов и входящих в их состав гликозаминогликанов может приводить к некоторым системным костным дисплазиям. Что касается деградации гликозаминогликанов, то в настоящее время известно свыше 10 типов лизосомных болезней накопления гликозаминогликанов, от носящихся к группе мукополисахаридов и маркированных первич ными биохимическими дефектами гликаногидролаз. При мукополисахаридозах достаточно изучен основной ферментный дефект для каждого из типов заболевания, обусловливающий нарушение дегра дации гликозаминогликанов и накопление нерасщепившихся их в соединительнотканных клетках и строме различных органов и си стем. В соответствии с этим разработаны методы пренатальной диагностики. В то же время пути нарушения биосинтеза и процес синга протеогликанов при наследственных заболеваниях скелета изучены мало.
и
У больных ПАХ выявлено накопление в цистернах зернистой эндоплазматической сети хондроцитов неколлагенового белка, которое сочеталось с отсутствием нормальной формы протеогликанов при элек трофорезе хряща. Накапливающийся материал взаимодействовал с ан тителами к стержневому белку протеогликанов. Это позволило пред положить нарушение транспорта аномального стержневого белка к пластинчатому комплексу (аппарат Гольджи).
Совершенно новые возможности для клинической генетики в ортопедии были получены, когда в круг исследований был введен новый объект — соматические клетки человека, культивируемые in vitro. Культивированные клетки сохраняют генетическую информацию исходного организма, и в то же время благодаря выве дению клеток из организма исследователь получает возможность изучать разные аспекты жизнедеятельности клеток конкретного больного в контролируемых условиях. Поэтому использование культивируемых клеток представляется уникальным для изу чения патогенеза наследственных болезней и для выявления наследственно обуслов ленной изменчивости клеточных функций, предрасполагающей к развитию патоло гических изменений. Было сформулировано новое направление клинической гене тики-— клеточная генетика, где соматическая клетка выступает как самостоятельная биологическая единица, участвующая в передаче генетической информации; где можно, получив потомство отдельно взятой клетки, выявлять новые закономерности, изучение которых не может быть проведено на других уровнях и объектах.
Предмет клеточной генетики состоит в изучении генетического контроля основных клеточных функций и процессов, лежащих в основе организма и поддержания его жизнедеятельности: пролиферации, дифференцировки, локомоции, рецепции, т. е. в изучении эффекта мутантных генов на уровне клетки, поскольку именно клетка
иклеточные системы являются тем специальным аппаратом, который интегрирует многочисленные и сложные метаболические процессы, представляя собой структур но-топографическую основу взаимодействия генов. При этом культивирование кле ток — не просто технический прием; оно может считаться методом, доказывающим наследственную природу болезни, поскольку, если клетки, выделенные из организма, сохраняют в ряду клеточных поколений in vitro рассматриваемый признак, харак теризующий данную патологию, то это свидетельствует о наследственном ее характере. В ортопедии есть ряд проблем, к которым должен быть применен клеточно-генети- ческий подход. Основными тканями, входящими в состав опорно-двигательного ап парата, являются соединительная ткань и ее специализированные типы — костная
ихрящевая. Основными клеточными элементами этих тканей являются соответственно фибробласты, а также остеогенные и хрящевые клетки. Эти клеточные элементы в настоящее время могут быть выделены в культуру в чистом виде. В культу рал ьных условиях эти клетки не только размножаются, но и претерпевают дифференцировку, сохраняя морфологические особенности, присущие данным клеткам. Известно, что такие процессы, как воспаление, регенерация, дистрофии, дисплазии, осуществляются при кооперативном взаимодействии клеток. Выраженность, интенсивность, скорость этих процессов, переход за рамки физиологической нормы подвержены межинди видуальной изменчивости, которая во многом генетически детерминирована. Исходя из этого, можно полагать, что многие наследственные болезни опорно-двигательного аппарата представляют собой следствие генетически детерминированных нарушений морфогенеза. Необходимо учитывать, что процессы морфогенеза имеют место и в постнатальном периоде, и явления регенерации, заживления ран, воспаления могут также рассматриваться как проявления постнатального морфогенеза. Клиницистам хорошо известна индивидуальная изменчивость, проявляющаяся в характере течения воспалительного или регенерационного процесса. Показано, что определенная и стабильная частота лиц с неблагоприятным течением послеоперационных ран является проявлением иммунобиологического полиморфизма человека, который в значительной мере является наследственным. Клеточная генетика и здесь может внести опреде ленный вклад, поскольку практически все »участники» регенерационного процесса— фибробласты и макрофаги — могут быть исследованы в клеточных культурах. Таким образом, программа целенаправленных исследований культивируемых клеток при наследственных заболеваниях скелета и аномальном течении раневого процесса пре-
12
дусматривает изучение любых параметров жизнедеятельности клеток (структурнофункциональных, физиологических, биохимических), а также поведения клеточных сообществ в условиях культивирования. В перспективе это может создать предпосылки для прогнозирования развития патологического процесса или течения раневого про цесса.
Обобщая выдвигаемую концепцию о перспективах разработки клеточно-гене- тических подходов в клинике травматологии и ортопедии, уместно подчеркнуть, что исследование культуры фибробластов, возможно, является одной из наиболее обещающих методик для изучения наследственных болезней соединительной ткани.
Изучение мультифакториальных заболеваний открывает широкие перспективы дальнейшего прогресса наших знаний патогенеза большой группы заболеваний опор но-двигательного аппарата, таких как идиопатический (диспластический) сколиоз, врожденный вывих бедра, врожденная косолапость, остеохондроз и другие распро страненные аномалии развития костно-суставного аппарата. Полученные в лабора тории генетики ЦИТО оценки количественного вклада генетических и средовых факторов в этиопатогенез мультифакториальных заболеваний свидетельствуют о том, что их возникновение обусловлено совместным действием этих двух групп факторов. При этом генетический груз, сопровождаемый непрерывным воздействием средовых факторов, обусловливает преодоление так называемой границы (барьера) проявляе мое™, и у индивидуума — скрытого носителя генетической предрасположенности — болезнь проявляется. В этом плане огромное значение для практического здравоох ранения имеет надежное определение круга лиц, подверженных высокому риску заболеваний ввиду их наследственной предрасположенности. Эта группа лиц должна находиться под пристальным вниманием врачей (например, при диспансерном на блюдении) для проведения эффективных профилактических мероприятий. При этом важным вопросом является определение генетических ассоциаций врожденных по роков развития, так как эти данные могут углубить представления о взаимосвязи отдельных мультифакториальных заболеваний и быть использованы при определении групп риска в конкретных семьях и популяциях. Выявление лиц с высоким риском заболеваний должно стать краеугольным камнем в системе профилактики в ортопедии.
Одной из конечных целей генетических исследований в травматолого-ортопе- дической клинике является медико-генетическое консультирование. Семьи, нуж дающиеся в этом, можно условно разделить на три основные группы: 1) родители здоровы, но имеют ребенка с наследственным заболеванием и хотят выяснить вероятность повторения заболевания у последующих детей; 2) один из родителей болен; 3) родители здоровы, но имеют родственников, страдающих наследственным заболеванием или пороком развития.
Медико-генетическое консультирование можно с уверенностью отнести к очень сложной и ответственной области медицинской деятельности, осуществляющей первичную профилактику наследственных заболеваний. Перед консультантом не редко возникают сложные проблемы морального и этического характера. В этих сложных ситуациях совет врача может оказать косвенное влияние на благополучие семьи, в связи с чем особенно велика ответственность консультанта как за содер жание, так и за форму совета.
При определении прогноза потомства наиболее перспективным методом является пренатальная диагностика, позволяющая не прогнозировать рождение ребенка с болезнью, а диагностировать заболевание у плода. Так, при мукополисахаридозе (МПС) IH (болезнь Гурлер), МПС III (болезнь Санфилиппо), МПС VI (болезнь Марото — Лами) амниоцентез с последующим изучением клеток амниотической жидкости позволяет выявить пораженные плоды, а также обнаружить среди плодов гетерозигот по этим нарушениям. В связи с успехами в молекулярно-генетических исследованиях, касающихся несовершенного остеогенеза, перспективной в самое ближайшее время представляется пренатальная диагностика этого заболевания. При медико-генетическом консультировании других форм заболеваний скелета в их пренатальной диагностике весьма ценные результаты может дать комбинированная фетоамниография (в частности, при ахондроплазии).
Таким образом, в настоящее время не вызывает сомнений не обходимость применения генетических подходов к изучению био логических основ практически всех групп заболеваний человека, в
13
том числе относящихся к опорно-двигательному аппарату. Генетика входит в число тех фундаментальных наук, достижения которых все шире используются в травматологии и ортопедии и обеспечивают решение как теоретических, так и прикладных задач, таких как исследование патогенеза, разработка методов диагностики, профи лактики и лечения заболеваний.
1.2.КЛАССИФИКАЦИЯ
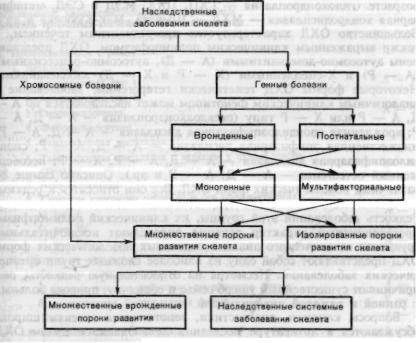
Клинические и генетические классификации наследственных забо леваний скелета (НЗС) весьма условны. НЗС можно разделить на хромосомные и генные. Действительно, большинство хромосомных болезней сопровождается тяжелыми множественными пороками раз вития скелета (синдромы Дауна, Эдвардса, Шерешевского — Тер нера, Клайнфелтера, Патау и др.), но не наоборот, так как далеко не все НЗС сопряжены с хромосомными аберрациями. Однако, как видно из исследований последних лет, граница между хромосомными и генными НЗС является условной и постепенно стирается по мере совершенствования методов генетического анализа. Так, метод ана лиза прометафазных хромосом, разработанный в последние десяти летия, озволил обнаружить микрохромосомные мутации при НЗС, которые ранее относили к генным. В частности, при трихоринофалангеальной дисплазии II типа (ТРФ-П, синдром Лангера — Гидиона — СЛГ), соединившей в себе фенотип ТРФ-I и множественные костно-хрящевые экзостозы, выявлена интерстициальная делеция длинного плеча хромосомы 8 в участке 8 (q 24,11 — q 24,13). Аналогичные перестройки обнаружены у части больных с ТРФ-1. Генные НЗС могут проявляться с момента рождения (ахондроплазия — АХ, диастрофическая дисплазия, врожденная косолапость и др.) или постнатально, когда ребенок рождается без видимых костных аномалий, заболевание развивается постепенно и манифестирует иногда лишь в последующие годы жизни — так называемый прогредиентный характер течения заболевания, обусловленный непре рывным действием продуктов функционирования мутантных генов (спондилоэпифизарная дисплазия — СЭД и МЭД, ПАХ и др.). По количеству вовлеченных локусов генные НЗС делят на менделирующие, в основе которых лежит действие главного гена (все 4юрмы наследственных остеохондродисплазий, многие 4юрмы изолирован ных пороков развития), и полигенные (мультифакториальные), к числу которых относят многие 4>ормы распространенных ортопеди ческих заболеваний (врожденный вывих бедра, остеохондроз позво ночника, диспластическии сколиоз и др.). НЗС также могут быть представлены изолированными ^юрмами и заболеваниями с множе ственным поражением скелета; среди последних различают множе ственные врожденные пороки развития (МВПР) и наследственные системные костные заболевания (НСКЗ) или собственно генерали зованные костные дисплазии. Условность генетической системати зации НЗС видна на схеме 1. Кроме упомянутого выше постепенного размывания грани между хромосомными и генными НЗС, условность
14
С х е м а 1. Генетическая систематизация наследственных заболеваний скелета
выражается также в том, что среди МВПР могут быть как хромо сомные, так и генные (менделирующие) формы, а среди изолиро ванных пороков развития — моногенные и мультифакториальные заболевания. Среди НЗС особое место занимают НСЗС, а среди последних — ОХД. Эта тяжелейшая категория наследственных за болеваний составляет обширную группу. ОХД относят к генерали зованным заболеваниям соединительной ткани, в связи с чем в патологический процесс могут быть вовлечены кость, сухожилия, связки, соединительнотканная основа глаза, внутреннего уха, внут ренних органов, хотя ведущим является поражение кости и хряща, что выражается в нарушении роста и развития костей и суставов. Многие формы ОХД сопровождаются непропорциональной карли ковостью: ризомелическое, мезомелическое, акромелическое укоро чение конечностей (АХ, мезомелическая дисплазия — ММД — типа Нивергельта, Лангера) либо укорочение туловища за счет преиму щественного поражения позвоночника (метатропическая дисплазия, дисплазия Книста, врожденная спондилоэпифизарная дисплазия — ВСЭД). Часть ОХД может выявляться при рождении и при этом представлять собой формы, летальные до или вскоре после рождения (ахондрогенез тип I и II), танатофорная дисплазия — ТД, синдром коротких бедер с полидактилией или без нее и др.), или оказываются совместимыми с жизнью (АХ, диастрофическая дисплазия — ДД,
15
метатропическая дисплазия, группа ММД и др.), вызывая тяжелые уродства. Значительная часть ОХД манифестирует в 3—6-летнем возрасте (гипохондроплазия — ГХП, ПАХ, МЭД и СЭД, метафизарная хондродисплазия — МХД — Шмидта, Мак-Кьюсика и др.). Большинство ОХД характеризуется прогредиентным течением, а также выраженным клиническим полиморфизмом. ОХД представ лены аутосомно-доминантными (А — Д), аутосомно-рецессивными (А — Р) и Х-сцепленными (X — Р и X — Д) заболеваниями. Некоторые формы ОХД генетически гетерогенны: заболевание с аналогичным клиническим фенотипом может наследоваться по А — Д, А — Р или X — Р типу (псевдоахондроплазия — А — Д, А — Р; врожденная спондилоэпифизарная дисплазия — А — Д, А — Р; множественная эпифизарная дисплазия — А — Д, А — Р; спон дилоэпифизарная дисплазия — А — Д,А — Р, X — Р; несовер шенный остеогенез — А — Д,А — Р и др.). Описано свыше 80 различных нозологических форм ОХД. Все они относятся к редким, но чрезвычайно тяжелым генерализованным заболеваниям скелета. Редкость заболеваний этой группы, их клинический полиморфизм, прогредиентныи характер течения определяют исключительные трудности генетического анализа отдельных нозологических форм. ОХД представляют собой одну из наиболее сложных групп ортопе дических заболеваний. Несмотря на относительную редкость, они причиняют существенный ущерб семье и обществу, приводя больных к ранней и тяжелой инвалидности.
Вопросы клиники, диагностики, генетические аспекты широко обсуждаются в литературе последних лет. Однако в целом ОХД остаются мало изученными и во многом спорными в группе на следственных заболеваний. Современная международная класси фикация ОХД уточняется и изменяется на систематически про водимых в Париже международных симпозиумах по вопросам номенклатуры и классификации болезней костей. Результаты ра боты этих симпозиумов опубликованы в различных периодических изданиях.
Согласно современной международной клинической классифика ции 1983 г., НСЗС подразделяются на группы ОХД, дизостозов, идиопатических остеолизов и первичных метаболических наруше ний. Однако это подразделение условно, учитывая отсутствие све дений о первичных молекулярных дефектах для большинства но зологических форм НСЗС. Современная клиническая классификация НСЗС может основываться лишь на данных клинико-рентгенологи- ческой и генеалогической интерпретации заболеваний.
Классификация в какой-то степени является схематичной, хотя она позволяет исследователям ориентироваться на единую терми нологию НСЗС.
Остановимся на основных формах ОХД, наиболее часто встре чающихся в практике ортопедов, педиатров и медицинских генети ков. Некоторые заболевания — юношеский эпифизеолиз головки бедренной кости и болезнь Эрлахера — Блаунта — изложены ниже (см. разделы 6.2 и 6.6).
16
КЛАССИФИКАЦИЯ НАСЛЕДСТВЕННЫХ СИСТЕМНЫХ ЗАБОЛЕВАНИЙ СКЕЛЕТА
Заболевания, характеризующиеся нарушением развития трубчатых костей и/или позвоночника
А.Проявляющиеся с рождения: I. Летальные:
1. |
Ахондрогенез тип I. |
А — Р |
2. |
Ахондрогенез тип П. |
|
3. |
Гипохондрогенез. |
А — Р |
4.Фиброхондрогенез.
5.Танатофорная дисплазия.
6.Танатофорная дисплазия с черепом в форме трили стника.
7.Ателоостеогенез.
8.Синдром короткого ребра (с и без полидактилии):
а) тип I; |
А — Р |
|
б) |
тип II; |
А — Р |
в) |
тип III. |
А — Р |
П.Обычно не летальные:
1.Точечная хондродисплазия:
|
а) ризомелическая аутосомно-рецессивная форма; |
А — Р |
||
|
б) доминантная Х-сцепленная форма. |
X — Д |
||
2. Кампомелическая дисплазия. |
А — Р |
|||
3. Кифомелическая дисплазия. |
А — Д |
|||
4. Ахондроплазия. |
|
А — Д |
||
5. |
Диастрофическая дисплазия. |
А — Р |
||
6. Хондроэктодермальная дисплазия. |
А — Р |
|||
7. |
Асфиксирующая дисплазия грудной клетки. |
А — Р |
||
8. Метатропичесхая дисплазия. |
А — Д, А — Р |
|||
9. Врожденная спондилоэпифизарная дисплазия: |
|
|||
|
а) аутосомно-доминантная форма; |
А — Д |
||
|
б) аутосомно-рецессивная форма. |
А — Р |
||
10. |
Дисплазия Книста. |
|
А — Д |
|
11. Диссегментарная дисплазия. |
А — Р |
|||
12. |
Мезомелическая дисплазия: |
|
||
|
а) |
тип Нивергельта; |
|
А — Д |
|
б) тип Лангера; |
|
А — Р |
|
|
в) тип Рейнхардта; |
|
А — Д |
|
|
г) |
тип Роббинова; |
|
|
|
д) другие Типы. |
|
А — Р |
|
13. |
Акромезомелическая дисплазия. |
А — Р |
||
14. |
Черепно-ключичная дисплазия. |
А — Д |
||
15. |
Отопалатодигитальный |
синдром: |
|
|
|
а) |
тип I; |
|
X — Д |
|
б> |
тип II. |
|
X — Р |
16. |
Синдром |
Ларсена. |
А — Д, А — Р |
|
17. Другие синдромы множественных вывихов. |
|
А — F |
|||
Б. Проявляющиеся не с рождения: |
|
|
|
|
|
1. Гипохондроплазия. |
|
|
|
А |
— Д |
2. Дисхондростеоз. |
|
|
|
А |
— Д |
3. Метафизарная хоидродисплазия типа Янсена. |
А — Д |
||||
4. Метафизарная хоидродисплазия типа Шмидта. |
А — Д |
||||
5. Метафизарная хоидродисплазия типа Мак-Кьюсика. |
А — Р |
||||
6. Метафизарная хоидродисплазия с экзокринной недоста- |
А — Р |
||||
точностью поджелудочной железы и циклической ней- |
|
|
|||
тропенией. |
|
|
|
|
|
7. Спондилометафизарная дисплазия: |
|
|
|
|
|
а) тип Козловского; |
|
|
|
А — Д |
|
б) другие типы. |
|
|
|
|
|
8- Множественная эпифизарная дисплазия: |
|
|
|
|
|
а) тип Фейрбанка; |
|
|
|
А — Д |
|
б) ругие формы. |
|
|
|
|
|
9. Множественная эпифизарная дисплазия |
с |
ранним |
А — Р |
||
диабетом. |
|
|
|
|
|
10. Артроофтальмопатия. |
|
|
|
А — Р |
|
11. Псевдоахондроплазия: |
|
|
|
|
|
а) доминантная форма; |
|
|
|
А — Д |
|
б) ецессивная форма. |
|
|
|
А — Р |
|
12. Поздняя спондилоэпифизарная дисплазия. |
|
|
X — Р |
||
13. Прогрессирующая псевдоревматоидная хондродиспла- |
А — Р |
||||
зия. |
|
|
|
|
|
14. Другие формы спондилоэпифизарной дисплазии. |
А — Р |
||||
15. Брахиолмия: |
|
|
|
|
|
а) аутосомно-рецессивная форма; |
|
|
А — Р |
||
б) аутосомно-доминантная форма. |
|
|
А |
— Д |
|
16. Дисплазия Диггве — Мельхиора — Клаузена. |
А — Р |
||||
17. Спондилоэпиметафизарная дисплазия (тяжелая фор |
|
|
|||
ма). |
|
|
|
|
|
18. Спондилоэпиметафизарная |
дисплазия |
с |
вывихами |
А — Р |
|
суставов. |
|
|
|
|
|
19. Отоспондиломегаэпифизарная дисплазия. |
|
|
А — Р |
||
20. Миотоническая хоидродисплазия. |
|
|
А — Р |
||
21. Парастрематическая дисплазия. |
|
|
А — Д |
||
22. Трихоринофалангеальная дисплазия. |
|
|
А — Д |
||
23. Акродисплазия с пигментацией сетчатки и нефропа- |
А — Р |
||||
тией. |
|
|
|
|
|
Заболевания, характеризующиеся |
нарушением |
|
|
|
|
развития хряща и фиброзного компонента скелета |
* |
|
|||
1. Гемимелическая эпифизарная дисплазия. |
|
|
|
|
|
2. Множественные хрящевые экзостозы. |
|
|
А — Д |
||
3.Энходроматоз (болезнь Оллье).
4.Акродисплазия с экзостозами (синдром Лангера—Гиди- она).
18
5. Энхондроматоз с гемангиомой (синдром Маффучи). |
|
|
6. |
Метахондроматоз. |
А — Д |
7. |
Спондилоэнхондроплазия. |
А — Р |
8.Остеоглофоническая дисплазия.
9.Фиброзная дисплазия.
10. |
Фиброзная дисплазия с гиперпигментацией кожи (син- |
А — Д |
|
|
дром Олбрайта). |
|
|
11. |
Семейная фиброзная дисплазия нижней челюсти. |
А — Д |
|
Заболевания, характеризующиеся нарушением |
|
|
|
толщины кортикального слоя диафиза и/или |
|
|
|
моделирования метафизов |
|
|
|
1. Несовершенный остеогенез. |
А — Р, А — Д |
||
2. Ювенильный идиопатический остеопороз. |
|
|
|
3. |
Остеопороз с псевдоглиомой. |
А — Р |
|
4. |
Остеопетроз: |
|
|
|
а) аутосомно-рецессивная летальная форма; |
А — Р |
|
|
б) промежуточная рецессивная форма; |
А — Р |
|
|
в) рецессивная форма с тубулярным ацидозом; |
А — Р |
|
|
г) аутосомно-доминантная форма. |
А — Д |
|
5. Пикнодизостоз. |
А — Р |
||
6. |
Доминантный остеосклероз типа Станеску. |
А — Д |
|
7. |
Остеомезопикноз. |
А — Д |
|
8. |
Остеопойкилия. |
А — Д |
|
9. |
Полосатая остеопатия. |
А |
— Д |
10. |
Полосатая остеопатия + склероз черепа. |
А |
— Д |
11. Мелореостоз. |
|
|
|
12. |
Диафизарная дисплазия Энгельманна — Камурати. |
А — Д |
|
13.Черепно-диафизарная дисплазия.
14.Эндостальный гиперостоз:
|
а) аутосомно-доминантная форма; |
А — Д |
|
б) аутосомно-рецессивная форма; |
А — Р |
|
в) аутосомно-рецессивная форма (склероостеоз). |
А — Р |
15. |
Тубулярный стеноз. |
А — Д |
16. |
Пахидермопериостоз. |
А — Д |
17.«Остеодисплазия Мельника — Нидльса. |
А — Д |
|
18. |
Фронтометафизарная дисплазия. |
X — Р |
19. |
Краниометафизарная дисплазия. |
А — Д |
20. Метафизарная дисплазия (болезнь Пайла). |
А — Д или А — Р |
|
21. |
Дизостеосклероз. |
А — Р или X — Р |
22. |
Остеоэктазия с гиперфосфатазией. |
А — Р |
23. |
Глазозубокостная дисплазия: |
|
|
а) легкая форма; |
А — Д |
|
б) тяжелая форма. |
А — Р |
24. |
Младенческий кортикальный гиперостоз. |
А — Д |
Особые трудности в разногласиях в генетическом анализе связаны с диагностикой группы костных дисплазий с поражением эпифизов. Поражение эпифизов сопутствует многим формам системных кост ных заболеваний. При накоплении материала выяснилось, что су ществует целый ряд заболеваний, основное сходство которых со спондилоэпифизарной дисплазией заключается в самом факте по ражения эпифизов и позвоночника. При этом не учитывались ни характер поражения, ни его сочетание с другими скелетными и внескелетными изменениями. Наиболее типичными представителя ми эпифизарных дисплазий являются СЭД и МЭД.
1.3. ВИДЫ ЗАБОЛЕВАНИЙ
1.3.1.Спондилоэпифизарная дисплазия
Воснове СЭД лежит дефект развития суставного хряща. Этот дефект распространяется как на трубчатые кости, так и на позвоночник. Клиническая картина СЭД довольно вариабельна и во всех случаях проявляется по мере роста ребенка. Характерными для всех больных являются небольшой рост, быстрая утомляемость, боли в нижних конечностях при нагрузке.
Диагностируется это заболевание в большинстве случаев после того, как у ребенка появятся боли в суставах и деформации нижних конечностей. Такие дети в возрасте 1—2 лет ходят раскачиваясь. Они менее подвижны, чем их сверстники, быстро устают при ходьбе. На рентгенограмме тазобедренных суставов отмечаются очень ма леньких размеров ядра окостенения головок бедер.
В возрасте 3—6 лет у некоторых больных могут появляться непостоянные боли в суставах нижних конечностей. Движения в них обычно свободны, и только в некоторых случаях может быть незначительная болезненность при ротационных движениях в тазо бедренном суставе. Однако по такой клинической картине установить диагноз не представляется возможным. Поэтому основная роль в диагностике СЭД отводится рентгенологическому исследованию. На рентгенограмах отмечаются изменения эпифизов всех трубчатых костей, но наиболее отчетливо бедренных костей и костей голени: эпифизы утолщены, имеют грибовидную форму. В области коленных суставов отмечаются сглаженность межмыщелковых возвышений болыпеберцовой кости и расширение межмыщелковой ямки бедрен ной кости, дольчатый надколенник (рис. 1.3). Все тела позвонков уплощены в той или иной степени, больше в грудном отделе.
У детей старше 8 лет нагрузка на нижние конечности постепенно увеличивается и в наиболее нагружаемых суставах (тазобедренных) прогрессируют изменения эпифизов и появляются первые клиниче ские признаки раннего деформирующего артроза. Вначале боли возникают при ходьбе, а затем и в покое. Характерным признаком начинающегося артроза является то, что ребенок после покоя сразу не может идти и первые шаги делает с большим трудом. Движения в этих суставах вначале мало изменены, но постепенно ограничение
20