

Полезные материалы за все 6 курсов / Учебники, методички, pdf / Шапошников_Травматология_и_ортопедия_3_том
.pdfлегче, так как отмечается отставание психомоторного развития. Отмечаются позднее закрытие родничка и запаздывание появления молочных зубов. Могут наблюдаться нефиксированный кифоз и ограничение отведения бедер.
Все точки окостенения, выявляемые рентгенологически, появля ются со значительным опозданием, но порядок их возникновения обычен.
Ранняя диагностика гипотиреоза необходима для начала свое временного лечения, так как дети, которые получают лечение с первых 6 мес жизни, могут нормально развиваться, чего нельзя сказать о детях, которым лечение начато поздно.
МПС II по картине костных изменений похож на МПС IH. Болеют всегда мальчики, большей частью со светлыми волосами и темными густыми бровями. Характерно для этого типа МПС исчез новение координации движений к 2—6 годам (ребенок часто падает), поведение детей становится неровным, а иногда и агрессивным. Они плохо едят твердую пищу. Между лопатками бывает узелковое поражение кожи. Помутнения роговицы обычно не наблюдается. Черты лица имеют нерезко выраженный Гурлер-подобный характер.
При МПС IS первые симптомы появляются поздно (в 3—6-летнем возрасте) и развиваются очень медленно. Полная клиническая кар тина выявляется к моменту половой зрелости. Помутнение роговицы обнаруживается в подростковом возрасте и часто более выражено по периферии. Гепатоспленомегалия бывает редко. Типичен для МПС IS дефект клапана аорты. Умственное развитие понижено незначительно, а иногда не страдает. Поражение костно-суставной системы по тяжести самое легкое из всех МПС Гурлер-подобного типа.
При МПС VI — синдроме Марото — Лами первые признаки заболевания появляются после 2-летнего возраста и к 7—9 годам становятся очень характерными. Эти больные достигают 145—155 см роста. Черты лица грубые, но не такие, как при МПС IH или II, контрактуры в суставах выражены, дети «скованы» при движениях. Характерно для этого типа МПС, что клиническая картина выражена довольно резко, но умственное развитие никогда не страдает. Эти два фенотипа МПС сопровождаются характерными изменениями скелета.
Клиника Моркио-подобных МПС резко отличается от таковой при Гурлер-подобной патологии. Больные — карлики, со специфи ческим лицом и коротким туловищем. Первые признаки заболевания появляются после 2-летнего возраста и быстро нарастают. Голова довольно большая, грубые черты лица (гипертелоризм, седловидная переносица, выступающая нижняя часть лица). Шея короткая, килевидная грудная клетка, у многих отмечается кифоз. Лопатки расположены высоко, деформированы предплечья, кисти мягкие. Увеличены суставы (лучезапястные, коленные и голеностопные). Это особенно заметно из-за пониженного питания. Нижние конеч ности искривлены (genu valgum), стопы распластаны. Дети быстро устают, походка быстро ухудшается, и многие перестают ходить
41
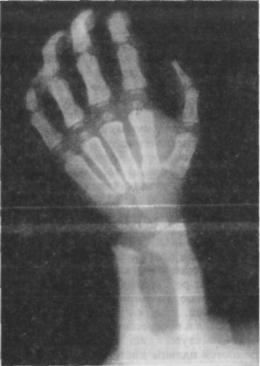
Рис 1.15. Рентгенограмма кисти. Мукополисахаридоз с Гурлер-подобным феноти пом (мукополисахаридоз VI типа).
Рентгенологически наиболее характерны изменения таза, тазо бедренных суставов и кистей. Таз сдавлен с боков, головки бедер маленькие, уплощены, имеется coxa valga, истончены шейки бед ренных костей. В кисти запаздывает появление ядер окостенения костей запястья, пястные кости короткие, широкие, проксимальные их отделы сужены, фаланги пальцев широкие, дистальные фаланги гипопластичны (рис. 1.15).
Тяжелее всех протекает МПС IH. Первые симптомы появляются в первые месяцы жизни ребенка и быстро прогрессируют. Наблю дается резкое снижение умственного развития. К 2—3 годам раз вивается типичная клиническая картина. Но надо помнить, что МПС IH на первом году жизни надо дифференцировать от врож денного гипотиреоза.
Дети с МПС IH часто рождаются с большей массой, у них могут быть затяжные желтухи. Характерны апатия, медлительность, от сутствие аппетита, запоры. Могут наблюдаться грыжи. У таких детей лицо одутловатое, язык большой и часто высунут, большой живот, сухая кожа. Во втором полугодии жизни диагноз установить
40
легче, так как отмечается отставание психомоторного развития. Отмечаются позднее закрытие родничка и запаздывание появления молочных зубов. Могут наблюдаться нефиксированный кифоз и ограничение отведения бедер.
Все точки окостенения, выявляемые рентгенологически, появля ются со значительным опозданием, но порядок их возникновения обычен.
Ранняя диагностика гипотиреоза необходима для начала свое временного лечения, так как дети, которые получают лечение с первых 6 мес жизни, могут нормально развиваться, чего нельзя сказать о детях, которым лечение начато поздно.
МПС II по картине костных изменений похож на МПС IH. Болеют всегда мальчики, большей частью со светлыми волосами и темными густыми бровями. Характерно для этого типа МПС исчез новение координации движений к 2—6 годам (ребенок часто падает), поведение детей становится неровным, а иногда и агрессивным. Они плохо едят твердую пищу. Между лопатками бывает узелковое поражение кожи. Помутнения роговицы обычно не наблюдается. Черты лица имеют нерезко выраженный Гурлер-подобный характер.
При МПС IS первые симптомы появляются поздно (в 3—6-летнем возрасте) и развиваются очень медленно. Полная клиническая кар тина выявляется к моменту половой зрелости. Помутнение роговицы обнаруживается в подростковом возрасте и часто более выражено по периферии. Гепатоспленомегалия бывает редко. Типичен для МПС IS дефект клапана аорты. Умственное развитие понижено незначительно, а иногда не страдает. Поражение костно-суставной системы по тяжести самое легкое из всех МПС Гурлер-подобного типа.
При МПС VI — синдроме Марото — Лами первые признаки заболевания появляются после 2-летнего возраста и к 7—9 годам становятся очень характерными. Эти больные достигают 145—155 см роста. Черты лица грубые, но не такие, как при МПС Ш или II, контрактуры в суставах выражены, дети «скованы» при движениях. Характерно для этого типа МПС, что клиническая картина выражена довольно резко, но умственное развитие никогда не страдает. Эти два фенотипа МПС сопровождаются характерными изменениями скелета.
Клиника Моркио-подобных МПС резко отличается от таковой при Гурлер-подобной патологии. Больные — карлики, со специфи ческим лицом и коротким туловищем. Первые признаки заболевания появляются после 2-летнего возраста и быстро нарастают. Голова довольно большая, грубые черты лица (гипертелоризм, седловидная переносица, выступающая нижняя часть лица). Шея короткая, килевидная грудная клетка, у многих отмечается кифоз. Лопатки расположены высоко, деформированы предплечья, кисти мягкие. Увеличены суставы (лучезапястные, коленные и голеностопные). Это особенно заметно из-за пониженного питания. Нижние конеч ности искривлены (genu valgum), стопы распластаны. Дети быстро устают, походка быстро ухудшается, и многие перестают ходить
41
(причиной является компрессия спинного мозга). Рентгенологически характерны гипоплазия или отсутствие зуба Си и признаки неста бильности атлантозатылочного сустава. Наблюдается универсальная платиноспондилия. Характерны также изменения таза: вертлужные впадины широкие, плоские, вдавленные в малый таз, крылья под вздошных костей изогнуты, нависают над головками бедренных костей. Шейки их вальгусные, головки резко уплощены, фрагментированы. Изменения в кистях похожи на изменения в Гурлер-по- добной группе.
Дети часто плохо слышат (ранняя тугоухость); отмечается склон ность к простудным заболеваниям. Нередко наблюдаются пупочные и/или паховые грыжи. Интеллект в большинстве случаев не снижен. Часто отмечаются изменения со стороны сердечно-сосудистой сис темы по типу кардиопатий, увеличение печени, реже — селезенки. Особенно резко выражены изменения со стороны глаз: характерны дистрофические изменения роговиц, чаще всего выявляемые с по мощью щелевой лампы. Суммарная экскреция гликозаминогликанов с мочой во всех случаях повышена. Обнаруживается также большое количество метахроматически окрашивающихся гранул в полимор фно-ядерных лейкоцитах и лимфоцитах больных.
Вместе с тем все МПС являются болезнями накопления глико заминогликанов, которое происходит за счет дефекта деятельности лизосомных гидролаз, осуществляющих свою функцию в лизосомах. Одним из информативных методов диагностики МПС является изу чение ультраструктуры клеток соединительной ткани больного. На иболее доступным является исследование биоптатов кожи. При этом в фибробластах кожи больных с Гурлер-подобным фенотипом мукополисахаридоза обнаруживают характерные изменения.
Таким образом, Гурлер-подобная группа МПС имеет определен ный клинико-рентгенологический фенотип, отмечаются накопление гликозаминогликанов в лизосомах, обнаруживаемое в фибробластах кожи, и усиленное выделение гликозаминогликанов с мочой. Что касается Моркио-подобной группы, то такой диагноз можно уста новить на основании клинических, рентгенологических данных и гиперэкскреции гликозаминогликанов с мочой, в то время как при знаки накопления их в фибробластах биоптатов кожи не определя ются, так как кератансульфат, дефект расщепления которого обус ловливает развитие этой группы МПС, не синтезируется и соответ ственно не может накапливаться кожными фибробластами. При этом характерное накопление гликозаминогликанов имеет место в других соединительнотканных клетках (хондроцитах, кератоцитах).
1.3.11. Ахондроплазия
Совсем недавно большинство непропорциональных карликов расце нивались как больные АХ. В настоящее время выделено множество форм карликовости.
АХ является наиболее изученным типом из всех форм наслед ственной карликовости с укороченными конечностями. Дети, стра-
42
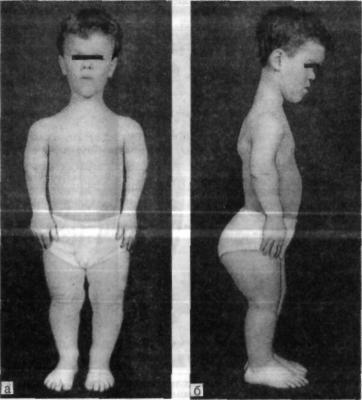
Рис. 1.16. Ребенок с ахондроплазией.
а — вид спереди; б — вид сбоку.
дающие истинной АХ, уже при рождении имеют характерную кли ническую картину: крупную голову, диспропорциональное телосло жение с ризомелическим укорочением конечностей. Голова может быть резко увеличена, с большой мозговой частью и с резко уве личенными родничками. Лицо с выпуклым лбом и уплощенной переносицей. Конечности укорочены, концы пальцев кистей при резком укорочении могут доходить до паховой складки, а при меньшем укорочении — до большого вертела или до верхней трети бедра (рис. 1.16). На плечах и бедрах наблюдаются глубокие кожные складки из-за избыточного количества мягких тканей. Кисти ко роткие, широкие, изодактилия, пальцы расположены в виде тре зубца. У детей первого года жизни отмечается разболтанность ко ленных суставов. Стопы короткие, широкие. Туловище обычно нор мальной величины, спина или прямая, или отмечается кифоз в грудопоясничном отделе позвоночника, который даже у маленьких детей фиксирован и плохо поддается коррекции.
43
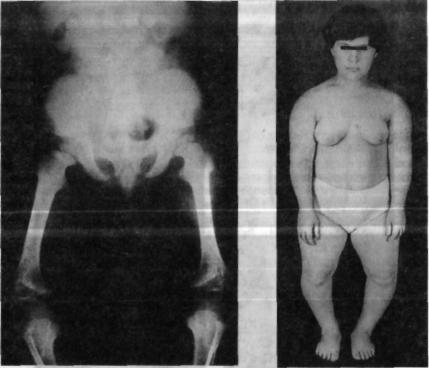
1
Рис. 1.17. Рентгенограмма тазобедренных и коленных суставов. Ахондроплазия.
Рис. 1.18. Общий вид больной с гипохондроплазией.
Дети с АХ отстают в моторном развитии, поздно садятся и встают на ноги. Но это не значит, что их надо приучать сидеть и ходить, так как, когда ребенка сажают, у него резко увеличивается кифоз, а ранняя ходьба может вызвать вальгусное искривление нижних конечностей. Когда ребенок начинает ходить, появляется усиление поясничного лордоза.
При рождении длина ребенка может быть нормальной (50—52 см) или меньше нормы (44—47 см). При диагностическом наблюдении за больными мы могли отметить, что дети, родившиеся с длиной меньше нормы, в дальнейшем имеют более выраженную диспропорцию тело сложения и очень маленький рост, поражения скелета у них были более тяжелыми. С ростом ребенка может появиться варусная деформация в коленных и голеностопных суставах, которая объясняется более бы стрым ростом малоберцовой кости.
Обычно больные АХ бывают практически здоровыми людьми, но у части из них (во взрослом состоянии) отмечается компрессия спинного мозга. У детей такие осложнения встречаются чрезвычайно редко. Такие дети должны быть под наблюдением нейрохирургов.
44
Диагноз АХ обычно устанавливают клинически, рентгенологи ческое исследование только подтверждает его. Патогномоничным рентгенологическим признаком является сужение расстояния между ножками дуг поясничных позвонков в каудальном направлении. Кроме этого, отмечаются развернутые и укороченные крылья под вздошных костей, крыши вертлужных впадин горизонтальны, по перечный размер входа в малый таз превышает его глубину. Труб чатые кости укорочены, метафизарные отделы их утолщены, чаше образно расширены (рис. 1.17).
1.3.12. Гипохондроплазия
ГХП в прошлом рассматривалась как «стертая» форма АХ (рис. 1.18). Основные дифференциально-диагностические различия АХ и ГХП представлены в табл. 4.
Т а б л и ца 4. Основные дифференциально-диагностические различия ахондро-
плазии и гипохондроплазии |
|
., |
|
|
|
|
|
Симптом |
Ахондроплазия |
Гипохондроплазия |
|
|
|
|
|
Появление первых при |
С рождения |
|
К 2—3 годам; наиболее чет |
знаков заболевания |
|
|
к о — к 7 годам |
Изменения со стороны: |
|
|
|
лица и головы |
Голова большая, с навис |
Голова и лицо нормальных |
|
|
шим лбом; седловидный |
размеров и пропорций |
|
|
нос |
|
|
|
Широкие, с короткими |
Широкие, пальцы располо |
|
кистей |
толстыми пальцами, рас |
жены обычно, иногда нерез |
|
|
положенными |
веером |
ко выраженная изодактилия |
|
и/или в виде |
трезубца; |
|
|
изодактилия |
|
|
|
Симптом сужения рас |
Расстояние между корнями |
|
позвоночника |
стояния между ножками |
дужек поясничных позвон |
|
|
дуг поясничных позвон |
ков в каудальном направле |
|
|
ков в каудальном на |
нии не суживается, как при |
|
|
правлении |
|
АХ, но и не расширяется, |
|
|
|
как в норме; оно остается |
|
|
|
примерно одинаковым |
|
|
|
|
1.3.13. Танатоформная дисплазия
ТД была описана P. Marofeaux в 1967 г., причем свое название она получила от греческого слова «танатос» (что означает «смерть») и «форус» (ищущий), так как больные дети рождаются мертвыми или умирают от респираторных заболеваний сразу или вскоре после рождения. Заболевание чаще всего смешивают с АХ. При рождении ТД характеризуется резким укорочением конечностей, туловищем относительно нормальной длины и довольно крупной головой с явно выраженной диспропорцией черепно-лицевого отдела по типу АХ. Отличительной особенностью ТД является сужение грудной клетки,
45
напоминающее грушу. Может иметь место тяжелая гидроцефалия, которая затрудняет рождение ребенка. У больных ТД были описаны самые разнообразные внескелетные аномалии: незаращение артери ального (боталлова) протока, атриосептальные дефекты, сужение стенок аорты, аномалии мозга. Рентгенологические признаки ТД являются диагностическими: резко выраженная платиспондилия с относительно расширенными межпозвоночными дисками; каудальное расширение позвоночного канала. Бедренные кости выглядят искривленными. Ребра укорочены и уплощены, грудина вдавлена.
1.3.14. Мезомелические дисплазии
ММД — это гетерогенная группа ОХД, для которой характерно укорочение прежде всего лучелоктевых и болынеберцовых и мало берцовых сегментов конечностей. ММД объединяют 6 типов забо левания, из которых 5 проявляются уже при рождении ребенка (типы Нивергельта, Лангера, Роббинова, Рейнхардта — Пфейффера и Вернера). Наиболее часто встречающийся тип ММД — дисхондростеоз — единственный представитель мезомелических дисплазии, проявляющийся не с рождения, а в основном в дошкольном возрасте. Наиболее часто проявляется умеренным снижением роста. Другие признаки — укорочение предплечья и голени — становятся замет ными более поздно. В отличие от других представителей остеохондродисплазий для дисхондростеоза типична лучевая косорукость (так называемая двусторонняя деформация Маделунга). На рентге нограмме определяются треугольная форма первого ряда костей запястья, подвывих или вывих кисти в лучезапястном суставе. Лу чевая кость укорочена и кисть смещена в тыльную сторону, вслед ствие чего верхняя конечность при осмотре сбоку напоминает «штык» (рис. 1.19). Дифференцировать дисхондростеоз следует от деформа ции Маделунга, которая бывает односторонней. В настоящее время общепризнано, что все случаи «двусторонней деформации Маделун га» следует рассматривать как дисхондростеоз.
ММД Нивергельта. Характеризуется низким ростом и укороче нием конечностей мезомелического типа, чаще нижних. Укорочение наблюдается в голенях, на медиальной или латеральной стороне которых имеется костный выступ. В коленных суставах вальгусная деформация. Стопы часто деформированы. При поражении верхних конечностей отмечаются короткие искривленные предплечья. Огра ничены супинация и разгибание в локтевых суставах. Кисть может иметь ульнарную девиацию и сгибательные контрактуры в пальцах. Рентгенологически: болыпеберцовая кость плоская, часто ромбовид ной формы, предплюсневые, плюсневые кости и фаланги пальцев могут синостозироваться. Кости предплечья короткие, изогнутые. Обычно бывает лучелоктевой синостоз. В наших наблюдениях были больные с поражением как нижних, так и верхних конечностей.
ММД Рейнгхардта — Пфейффера. Проявляется с рождения умеренной мезомелией. Отличается от других представителей ММД тем, что диафизы локтевой и малоберцовой костей плоские и ду-
46

Рис. 1.19. Деформация предплечья при дисхондростеозе.
гообразно искривлены. Дистальные отделы локтевой кости и про ксимальные отделы малоберцовых костей недоразвиты, головки лу чевых костей вывихнуты.
ММД Вернера. Проявляется с рождения диспропорциональной карликовостью с выраженной мезомелиеи голеней при почти полном отсутствии изменений в верхних конечностях. Иногда может отсут ствовать I палец на кисти и стопе, может определяться полидактилия. Рентгенологическая картина типична: болыиеберцовая кость руди ментарная, вследствие чего относительно интактная головка мало берцовой кости вывихнута.
ММД Лангера. Это проявляющаяся с рождения диспропорцио нальная карликовость с одинаково выраженной мезомелиеи верхней и нижней конечностей. На рентгенограммах определяются гипопла зия верхней трети лучевой кости, недоразвитие проксимальной части костей голени, рудиментарная малоберцовая кость.
ММД Роббиновг. Проявляется с рождения умеренной мезоме лиеи, сочетающейся с признаками лицевого дисморфизма —макро цефалией, выступающим лбом, широкой переносицей, гипертелоризмом, седловидным носом, треугольной формы ртом с открытым вниз углом. Предплечья укорочены сравнительно больше, чем го лени. Определяется вывих головок лучевых костей, но при этом разгибание в локтевых суставах полное. Характерны гиперподвиж-
47
ность в межфаланговых суставах кисти и стопы, брахидактилия. Патогномоничным признаком заболевания является гипоплазия на ружных половых органов.
1.3.15. Метафизарные хондродисплазии
Метафизарные дисплазии являются гетерогенной группой заболева ний, характеризующихся поражением преимущественно метафизов с относительно нормальными эпифизами и позвоночником. Наиболее ярким представителем метафизарных дисплазии является группа МХД. В основе МХД лежит задержка энхондрального роста, про исходящая вследствие недостаточного и неправильного окостенения в области метафизов длинных трубчатых костей. Развитие эпифизов и рост костей со стороны периоста не нарушаются. Клинически эта группа заболеваний характеризуется отставанием роста и деформа циями конечностей у больных.
Тип Янсена. Является наиболее тяжелым. Встречается очень редко. Все авторы при описании клинико-рентгенологической кар тины заболевания ссылаются на первое классическое наблюдение М. Yansen (1934). Внешний вид больного настолько характерен, что если врач один раз увидит такого пациента, то в следующий раз ему не придется дифференцировать это заболевание от других. Телосложение у таких больных непропорциональное, имеется от ставание в росте, череп башенный. Глаза широко расставлены, иногда наблюдается экзофтальм, переносица широкая и приплюс нутая, нижняя челюсть недоразвитая, отмечается неправильный прикус. Осанка сутулая, плечи повернуты вперед и как бы стянуты ключицами. Грудная клетка конусовидная, с выдающейся грудиной. Конечности несколько укорочены, искривлены, суставы увеличены
вобъеме, имеется варусное искривление локтевых суставов, кисти
столстыми пальцами в виде барабанных палочек. В тазобедренных
иколенных суставах имеются сгибательные контрактуры, голени саблевидные, стопы плосковальгусные. В результате таких дефор маций дети ходят на согнутых ногах; корпус отклонен вперед, руки висят спереди, часто доходят до колен и ниже. Сгибательные контр актуры вызывают у больного желание сидеть на корточках.
Рентгенологические изменения аналогичны во всех крупных су ставах: определяется неравномерная оссификация метафизов на зна чительном протяжении, очаги дефекта костной ткани на фоне из мененной структуры метафизов. Зоны роста резко расширены, кон туры эпифизов и метафизов неровные со стороны ростковой хря щевой пластинки. Длинные трубчатые кости укорочены и искривлены. С ростом больного структура костей восстанавливается, но сохраняются деформации. Позвоночник не изменен. Заболевание наследуется по аутосомно-доминантному типу.
Тип Шмидта. Наследуется по аутосомно-доминантному типу и характеризуется слабым или умеренным снижением роста, дефор мацией нижних конечностей (genu varum). Первые признаки забо левания часто появляются на втором году жизни (иногда и раньше):
48