

Полезные материалы за все 6 курсов / Учебники, методички, pdf / Шапошников_Травматология_и_ортопедия_3_том
.pdfТРАВМАТОЛОГИЯ И ОРТОПЕДИЯ
РУКОВОДСТВО ДЛЯ ВРАЧЕЙ
В 3 ТОМАХ
Под редакцией члена-корр. РАМН Ю. Г. ШАПОШНИКОВА
МОСКВА «МЕДИЦИНА» 1997
Щ4 \
ОРТОПЕДИЯ
томЗ
МОСКВА «МЕДИЦИНА» 1997
ББК 54.58
Т65 УДК 617.3(035)
Авторы: Е. А. Абальмасова, проф., Л. Н. Алякин, В. Л. Анд
рианов, проф., |
М. А. Берглезов, |
проф., А. П. Бережный, проф., |
||
B. Н. Бурдыгин, |
д-р |
мед. наук, |
А. Т. Бруско, д-р |
мед. наук, |
C. Т. Ветрилэ, проф., |
М. В. Волков, акад. РАМН, |
С. Е. Волков, |
||
Б. В. Гусев, доцент, С. Т. Зацепин, проф., И. С. Истомина, канд. мед. наук, А. Ф. Каптелин, проф., Н. И. Кондрашин, проф.,
A. А. Корж, акад. РАМН, проф., А. Ф. Краснов, акад. РАМН, B. Н. Кувина, д-р-мед. наук, О. А. Малахов, проф., Е. М. Меерсон, проф., В. Н. Меркулов, д-р мед. наук, И. М. Митбрейт, проф., И. А. Мовшович, проф., Е. А. Назаров, П. В. Новиков, канд. мед. наук, Т. К. Нурмаганбетов, А. П. Поздеев, А. А. Раззоков, д-р мед.
наук, |
С. С. Родионова, |
д-р |
мед. |
наук, А. С. Самков, канд. мед. |
наук, |
А. И. Снетков, |
д-р |
мед. |
наук, Г. М. Тер-Егиазаров, |
проф., Е. С. Тихоненков, проф., В. В. Троценко, д-р мед. наук, А. П. Чернов, Н. А. Шестерня, проф., И. Н. Шинкаренко, канд. мед. наук, Я. Б. Юдин, проф.
Травматология и ортопедия/Руководство для врачей: в Т 65 3 томах. Т. З/Под ред. Ю. Г. Шапошникова. — М.: Медицина,
1997. — 624 с: ил. ISBN 5-225-02668-0
В руководстве изложены диагностика и лечение больных с ортопеди ческой патологией. Дана информация по врожденной и системной патологии опорно-двигательного аппарата, дегенеративно-дистрофическим заболевани ям, остеохондропатиям, обменным заболеваниям, опухолям, а также по некоторым нозологическим единицам, занимающим особое место в ортопе дии: сколиозу, полиомиелиту,^ артритам, остеомиелиту и др.
Для травматологов, ортопедов.
„4108050000— 68 ^ |
- |
e ¥ W .. ,„ |
—039(01)—97— |
объявл. |
ББК 54.58 |
ISBN 5-225-02668-0 |
© Коллектив авторов, 1997 |
Г Л А В А 1
НАСЛЕДСТВЕННЫЕ СИСТЕМНЫЕ ЗАБОЛЕВАНИЯ СКЕЛЕТА
1.1.ГЕНЕТИЧЕСКИЕ АСПЕКТЫ ОРТОПЕДИЧЕСКОЙ ПАТОЛОГИИ
Впоследние десятилетия четко обозначилось выделение клинической генетики как науки, изучающей законы возникновения и развития наследственных дефектов костно-суставного аппарата. По методам и целям исследований эта область знания в значительной мере относится к медико-биологическим дисциплинам; выводы и реко мендации, вытекающие из работ по клинической генетике, тесно сопряжены с имеющими большое значение для здравоохранения прикладными проблемами борьбы с наследственной патологией и,
вчастности, с заболеваниями опорно-двигательного аппарата. Эта новая фаза развития ортопедии предусматривает изучение и при менение новейших достижений как клинической медицины, так и генетики. И хотя углубленные генетические исследования сегодня возможны лишь в условиях специализированных клиник и лабора торий, не вызывает сомнения необходимость ознакомления с про блемами генетики в ортопедии широкого круга врачей. От того, насколько серьезно и внимательно отнесутся практические врачи, осуществляющие первичный контакт с больным, к изучению •на следственных заболеваний опорно-двигательной системы, во многом зависят успехи в их профилактике и лечении.
Аппаратом наследственности являются хромосомы и расположён ные на них гены. Любой ген каждой из 46 хромосом человека (кроме половых хромосом у мужчин) имеет парный ген, располо женный в точно таком же участке (локусе) второй парной хромосомы и оказывающий влияние на тот же признак.
Один из аллелей генов вместе с несущей его хромосомой унас ледован от отца, а второй — от матери. Если из пары контрастных родительских признаков, контролируемых определенным геном, у потомков проявляется только один (он как бы доминирует над вторым), то такой признак и соответствующий аллель гена назы вается доминантным. Соответственно контрастный к первому при знак (и аллель гена), который расположен в том же локусе парной хромосомы и, переходя к потомству, не проявляется, носит название рецессивного. Это относится к генам, контролирующим как нор мальные, так и патологические признаки. Это положение справед-
5
ливо для всех пар хромосом, кроме половых (или, согласно гене тической терминологии, для аутосом). Тогда говорят об аутосомнодоминантных и аутосомно-рецессивных признаках. Отсюда происте кает возможность аутосомно-доминантного и аутосомно-рецессивно- го типа наследования патологии. При первом типе патологический признак передается от поколения к поколению, при втором может длительно существовать скрытое гетерозиготное носительство мутантного гена, а рецессивное наследственное заболевание может возникнуть в случае брака двух носителей в результате образования патологической гаметы.
Возможен еще один тип наследования: наследование, сцепленное с полом и наблюдаемое в тех случаях, когда мутантный ген рас положен на одной из половых хромосом — X или Y. Особенности этого типа наследования обусловлены непарностью половых хромо сом у мужчин (XY), в результате чего гены, локализованные в каждой из половых хромосом мужчины, также непарны. Практи чески речь идет о наследовании, сцепленном с Х-хромосомой, так как достоверных сведений о наследовании, сцепленном с Y-хромо- сомой человека, нет. Наследование, сцепленное с полом, также может быть доминантным и рецессивным, однако имеет свои ха рактерные признаки, четко выявляемые при изучении родословных.
В сложнейшем шифре наследственности под влиянием множества причин эндогенного и экзогенного характера (хронические или ин фекционные заболевания, интоксикации, повышенный возраст ро дителей, физические, химические, биологические воздействия и др.) нередко возникают изменения, ошибки-мутации. Мутации опреде ляются как изменение наследственного вещества (хромосомные му тации, генные мутации), которое вызывает новое, передающееся по наследству изменение в организме. Мутации могут происходить на уровне всего хромосомного комплекса (уменьшение или увеличение числа хромосом), на уровне отдельной хромосомы (утрата, приоб ретение или изменение участка хромосомы) и на уровне гена (на участке ДНК, кодирующем синтез одной полипептидной цепи). Последнюю категорию мутаций называют генными (или точковыми) мутациями. Именно они наиболее часто лежат в основе многочис ленных наследственных заболеваний. При оплодотворении мутантной половой клетки мутация переходит к следующему поколению, повторяясь в каждой клетке тела нового организма. В процессе его развития мутация повлечет за собой соответствующие отклонения в синтезе белковых молекул, а затем в строении тканей, органов и систем. Мутация может поразить любой участок аппарата на следственности, имеющий отношение к формированию любого ор гана, ткани, обмена. Поэтому так бесконечно многообразны формы наследственных страданий. Иными словами, ни одна область кли нической медицины в настоящее время не может избежать столк новения с наследственными заболеваниями, независимо от того, вызвана ли наследственная болезнь у индивидуума мутацией, воз никшей в половой клетке его родителя, или мутацией, которая передалась через несколько поколений; и в том и в другом случае
б
болезнь должна считаться наследственной, хотя в первом случае заболевание представляется «спорадическим», как бы не связанным с наследственной передачей, так как родители и все другие родст венники здоровы, в то время как во втором случае заболевание прослеживается в одном или более предыдущем поколении. Таким образом, отсутствие изучаемого заболевания у предков или других родственников ни в коей мере не противоречит наследственному характеру заболевания.
Костный скелет как высокоорганизованная соединительная ткань подвержен тяжелым наследственным заболеваниям. Это генетически детерминированные расстройства, при которых поражаются преиму щественно кость и хрящ. Большинство заболеваний скелета в той или иной форме являются наследственными.
В настоящее время описано свыше 150 различных нозологических форм наследственных системных заболеваний скелета (НСЗС), пред ставленных аутосомно-доминантными (А — Д), аутосомно-рецес- сивными (А — Р) и Х-сцепленными (X — Р и X — Д) заболеваниями. Некоторые формы НСЗС генетически гетерогенны: заболевание с аналогичным клиническим фенотипом может наследоваться по-раз ному в различных семьях (А — Д, А — Р или X — Р тип), что может свидетельствовать о неоднородности существующих выделен ных нозологических форм, каждая из которых несет в себе по существу различные заболевания. Это относится к псевдоахондроплазии (ПАХ), множественной эпифизарной дисплазии (МЭД), не совершенному остеогенезу (НО) и другим заболеваниям (рис. 1.1).
При наследственных заболеваниях один ген может влиять на большее число признаков. Это явление носит название плейотропии. Вместе с тем один и тот же признак может находиться под контролем нескольких разных генов, нарушение которых приводит к одному
итому же эффекту (генокопирование). Оба явления — плейотропия
игенокопирование — широко известны в генетике человека и должны учитываться при обследовании больного НСЗС. В то же время этиологический фактор (функционирование мутантного гена) при НСЗС действует постоянно. Этим объясняется тот факт, что НСЗС являются, как правило, непрерывно текущими, прогрессиру ющими заболеваниями. Прогредиентный характер заболеваний дик тует необходимость динамического наблюдения за больным.
Так, в раннем детстве многие из характерных симптомов забо левания не проявляются, а обнаруживаются позднее, и тогда синдром выявляется полностью. Поэтому отсутствие какого-либо признака заболевания в раннем детстве не противоречит диагнозу. При изу чении семей с НСЗС следует иметь в виду возможность неполной пенетрантности — отношение числа больных или пораженных к общему числу носителей гена болезни (рис. 1.2) и изменчивой экспрессивности патологического гена. Последняя выражается в су щественном полиморфизме НСЗС (разное время появления симп томов или начала заболевания; различная степень выраженности клинических проявлений: количества симптомов, их тяжести, прогредиентности заболевания и др.). Вариации в проявлении НСЗС,
7
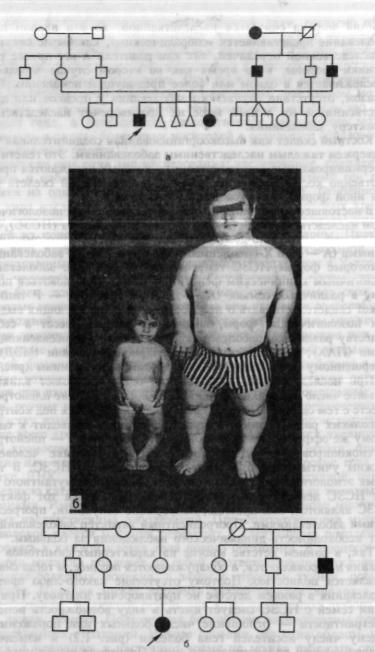
Рис. 1.1. Наследственные системные костные заболевания с разным типом насле дования.
а — родословные с аутосомно-доминантным типом наследования множественной экзостозной хондродисплазии; б — больные отец и дочь в семье с аутосомно-доминантным типом наследования псевдоахондродисплазии; в — больные отец, сын и дочь в семье с аутосом-
8
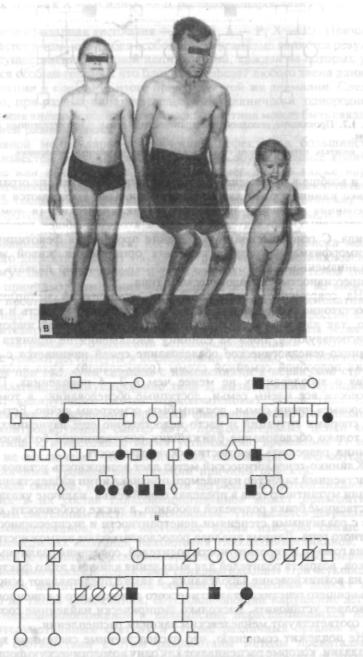
но-доминантным типом наследования множественной эпифизарной дисплазии; г — родословная с аутосомно-рецессивным типом наследования диастрофической дисплазии.
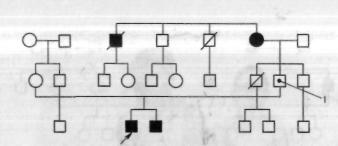
Рис. 1.2. Проявление неполной пенетрантности в семье с несовершенным остеогенезом.
1 — носитель патологического гена без клинических проявлений заболевания.
как и вообще в проявлении наследственных болезней, не ограничены только клиническими характеристиками. Они выражаются также в колебаниях значений различных параклинических (в том числе лабораторных) показателей, которые входят в общее понятие фе нотипа. С генетической точки зрения проявления фенотипического полиморфизма на разных ступенях организации живой материи (организменном, клеточном, субклеточном) можно назвать разной экспрессивностью патологического гена.
На современном этапе развития клинической медицины уже недостаточно обследования только самого больного, пусть и подроб ного, так как многие возможности получения ценной информации не используются, пока за единицу наблюдения не принята семья. Клинико-генеалогическое обследование семей начинается с состав ления подробной семейной схемы (родословной), куда входят све дения о заболеваниях не менее чем в 3—4 поколениях. По воз можности все члены семьи, доступные обследованию, в том числе «здоровые» члены семьи, должны быть осмотрены лично. Есть целый ряд спорных ситуаций и часто недостаточно еще изученных форм, где только обследование ближайших родственников больного в со стоянии разрешить диагностическую трудность.
Клинико-генеалогический метод дает возможность установить на следственный характер изучаемого признака и тип наследственной пе редачи мутантного гена в пределах одной семьи, наличие указаний на родственные браки родителей пробанда, а также особенности, связан ные с различными степенями пенетрантности и экспрессивности му тантного гена. Данные анализа родословных дают возможность выяс нения гомо- и гетерозиготности родителей, сочетания различных при знаков, возраста родителей для выявления влияния этого фактора ри ска на возникновение заболевания, а также представляют основу для дальнейшего генетико-статистического анализа, что в конечном счете позволяет установить, насколько эмпирически найденные соотноше ния соответствуют менделевским законам расщепления.
Не подлежит сомнению, что наследственные системные костные дисплазии, которые расценивают как одну нозологическую форму, мо гут быть обусловлены совершенно разными мутациями. Клинически сходное заболевание в одних семьях наследуется А — Р, в других —
10