

Лекция 11
Индуцированный мутагенез. Репарация генетического материала. Механизмы репарации ДНК. Понятие о генных болезнях.
В прошлой лекции шла речь главным образом о спонтанных (естественных) генных мутациях. Однако мутации могут быть индуцированными, возникать под действием ряда искусственных мутагенных факторов. Природа мутагенных факторов разнообразна, но в общем они делятся на физические, химические и биологические мутагенные факторы. По механизму возникающих изменений в генетическом материале между спонтанным и индуцированным мутагенезом принципиальных различий нет. Все факторы индуцированного мутагенеза будь они физическими, химическими или биологическими в конечном итоге нарушают молекулярную структуру отдельных генов, вызывают нарушения последовательности нуклеотидов с вытекающими отсюда последствиями.
Мутагенное действие физических факторов
К физическим мутагенным факторам относятся ионизирующее и ультрафиолетовое излучение. К ионизирующему излучению относятся электромагнитные колебания (рентгеновское и гамма-излучение) и потоки частиц (протоны, нейтроны, альфа-частицы и др.). Их энергия, проходя через клетку, вырывает электроны из внешней оболочки атомов или молекул, превращая их в нестабильные свободные радикалы и ионы. Свободные радикалы и ионы вступают в разнообразные химические реакции, что прямо или косвенно затрагивает азотистые основания в структуре ДНК и приводит к точечным мутациям. Мутагенный эффект ионизирующего излучения может быть связан с радиолизом воды в клетке. При этом образуются генотоксические радикалы и молекулы: гидроксильный радикал; супероксидный радикал.; перекись водорода. В тех случаях, когда энергия ионизирующих излучений поглощается непосредственно молекулами ДНК, прямо вызывая в них первичные повреждения, имеет место прямое воздействие радиации на генетический аппарат.
Один из видов электромагнитных колебаний является ультрафиолетовое излучение. В отличие от ионизирующего излучения ультрафиолетовое облучение обладает малой энергией и не способно вызывать ионизацию. Под действием ультрафиолетовых лучей в молекуле ДНК образуются пиримидиновые димеры (димеризация пиримидинов), обычно между остатками тимина, находящимися рядом в одной из цепей (рис. 2). Реже встречаются димеры между остатками цитозина, либо тимина и цитозина. Это следует рассматривать как предмутационное повреждение. Димеры нарушают спиральную конформацию ДНК и ингибируют нормальную репликацию, а это приводит к появлению ошибок репликации и развитию мутаций.
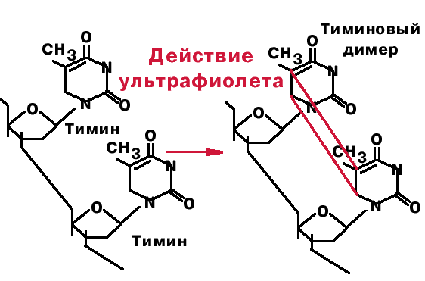
Рис. 2. Образование пиримидиновых димеров
Мутагенным действием обладает высокая температура. Мутагенный эффект повышенной температуры связан с ускорением процессов спонтанного мутирования. Это обеспечивает количественно небольшое влияние повышенной температуры на процессы мутаций. Уровень естественного мутирования повыщается в 2-3 раза при увеличении температуры на 10оС.
Мутагенное действие химических соединений
В настоящее время обнаружены сотни химических веществ, обладающих мутагенной активностью. Они имеются во всех классах химических соединений. Многие из них мутагенные в отношении как реплицирующейся, так и нереплицирующейся (покоящейся) ДНК: алкилирующие соединения, азотистая кислота. Однако есть мутагенные вещества, действующие на ДНК только в течение ее синтеза.
Обширная группа сильных химических мутагенов принадлежит к алкилирующим соединениям. Химически они являются источником введения в молекулы ДНК таких радикалов как метил (СН3-) - реакция метилирования ДНК, этил (С2Н5-) - реакция этилирования ДНК и другие. В качестве хорошо изученных мутагенов этой группы используют этиленимин, иприт, диэтилсульфат, нитрозоэтилмочевину и другие. Некоторые из них получили название супермутагенов за их исключительно высокую способность индуцировать мутации. Главное следствие мутагенного действия алкилирующих агентов – это появление мутаций типа транзиций ГЦ→АТ.
Сильным мутагенным действием обладает азотистая кислота (HNO2), действующая путем окислительного дезаминирования оснований, содержащих аминогруппы (гуанин, аденин, цитозин). Замещение аминогруппы кетогруппой превращает аденин в гипоксантин, спаривающийся преимущественно не с тимином, а с цитозином. Дезаминирование цитозина превращает его в урацил, спаривающийся с аденином. Все это ведет к генным мутациям по типу транзиций. Помимо замены оснований азотистая кислота индуцирует делеции оснований, что обусловлено ее способностью к поперечному сшиванию цепей ДНК.
Действие на клетку ряда химических мутагенов оказалось специфически приуроченным ко времени репликации ДНК. Это относится к аналогам оснований. Аналоги оснований - это соединения, имеющие сходную с нормальными азотистыми основаниями ДНК структуру, но отличающиеся от них по химическим свойствам. Это позволяет им вместо нормальных аналогов включаться в синтезируемые нити ДНК, что ведет в конечном итоге к мутационным последствиям типа транзиции.
Некоторые химические мутагены вызывают мутации сдвига рамки считывания, которые обусловлены добавлением к кодирующей последовательности или делецией одного, или нескольких нуклеотидов. Индукция таких мутаций была подробно исследована на примере акриловых красителей, относящихся к ароматическим углеводородам. К ним относятся, в частности профлавин и акридиновый оранжевый, молекулы которых близки по размерам к нуклеотидам и могут встраиваться между пиримидиновыми или пуриновыми нуклеотидами в молекуле ДНК. Это приводит к искривлению двойной спирали и появлению делеций или инсерций.
Мутагенное действие биологических объектов
К биологическим мутагенным агентам следует отнести мобильные генетические элементы, вирусы, токсические продукты жизнедеятельности паразитов. Они могут вызывать как генные, так и хромосомные мутации.
Ранее отмечалось, что развитие генных мутаций - это многоэтапный процесс, который начинается с первичных повреждений в молекуле ДНК, возникающих спонтанно или под действием особых факторов. Однако появление первичных повреждений еще не означает появление мутаций. Показано, что из общего числа первичных повреждений только часть их подвергается процессу фиксации мутаций, результатом которого служит: появление замены пар нуклеотидов, вставки или делеции пар нуклеотидов, их перестановки в ДНК. Большинство первичных повреждений подвергаются действию процессов репарации ДНК, направленных на восстановление целостности структуры молекулы. Благодаря системе репарации из 1000 повреждений ДНК различного типа лишь одно приводит к мутации. Репарация ДНК - это процесс ликвидации первичных повреждений в молекуле ДНК и замена их нормальными структурами. В репарации ДНК участвует более 150 разных генов. Механизмы репарации крайне важны для поддержания генетической целостности организмов и их выживания. В клетках про- и эукариот имеется много разных систем, ликвидирующих первичные повреждения. Из них можно выделить два основных типа репарации ДНК: дорепликативную и пострепликативную репарацию. Дорепликативная репарация включает фотореактивацию и различные формы темновой эксцизионной репарации.
Фотореактивация
Выше отмечалось, что облучение ультрафиолетом вызывает образование в молекулах ДНК пиримидиновых димеров. При этом образуются сшивки, как правило, между тиминами, расположенными рядом в одной нити ДНК. Восстановительный эффект при фотореактивации связан с действием фермента – фотолиазы (дезоксирибопиримидинфотолиазы). Фермент поглощает фотон света (синего спектра), после чего приобретает способность расщеплять ковалентные связи между остатками тимина в тиминовых димерах и восстанавливать поврежденную структуру ДНК. Такой механизм впервые был обнаружен у E. coli. в начале 60-х годов. Фермент фотореактивации в начале не был обнаружен у эукариот, которые используют другие механизмы репарации повреждений ДНК, индуцированных ультрафиолетом. Однако в дальнейшем фотореактивация была обнаружена в клетках многих эукариотических организмов. Появляются данные, указывающие на наличие фотореактивирующего фермента у млекопитающих, а также в клетках кожи человека.
Темновая эксцизионная репарация
В отличие от фотореактивации другие пути репарации ДНК не нуждаются в энергии видимого света и поэтому относятся к темновой репарации. Темновая эксцизионная репарация направлена на вырезание (эксцизию) участков ДНК, несущих повреждения с участием репарационных ферментов. Темновая эксцизионная репарация широко представлены как у прокариот, так и у эукариот и включает ряд этапов. Известно два типа эксцизионной репарации: репарация нуклеотидов и репарация оснований.
Репарация нуклеотидов
С помощью эксцизионной репарации нуклеотидов исправляются «блоки» ошибок, изменяющие структуру двойной спирали, например, пиримидиновые сшивки. Происходит это поэтапно с участием репарационных ферментов:
1. Фермент эндонуклеаза узнает повреждение в ДНК (повреждение может быть вызвано и димеризацией пиримидиновых оснований под влиянием ультрафиолетового излучения) и вносит одноцепочный разрыв фосфодиэфирной связи со стороны 5`-конца рядом с поврежденным участком.
2. 5`→3`-экзонуклеаза вырезает поврежденный участок, отщепляя нуклеотиды. После удаления участка с повреждением на этом месте образуется брешь.
3. Репарационная ДНК-полимераза заполняет эту брешь дезоксирибонуклеотидами, комплементарными нуклеотидам неповрежденной цепи ДНК, которые присоединяет с 3`-ОН конца.
4. ДНК-лигаза сшивает последний «разрез» цепи, который остается на 3`- ОН-конце последней пары оснований и закрывает брешь.
Репарация оснований
При эксцизионной репарации оснований исправляются повреждения азотистых оснований, вызванные спонтанно или химическими агентами. Первый этап такой репарации включает узнавание химически модифицированного основания ферментами ДНК-гликозилазами, которые специфичны для разных повреждений азотистых оснований ДНК. Этот фермент расщепляет связь между основанием и сахаром, удаляет поврежденное основание. Появляется апиримидиновый или апуриновый сайт (AП-сайт), который узнается АП-эндонуклеазой. Эндонуклеаза делает надрез на 5`-конце АП-сайта для образования 3`- ОН конца. ДНК-полимераза наращивает 3`- ОН конец и заполняет брешь, а ДНК-лигаза зашивает надрез. Следует отметить, что эукариотические ДНК-гликозилазы изучены намного хуже, чем гликозилазы прокариот. Кроме того, для человека и животных не создано модельных систем болезней с нарушенной эксцизионной репарацией оснований, поэтому оценить важность этого типа репарации довольно трудно.
Пострепликативная репарация
Известны и другие типы репарации, что говорит о большом разнообразии механизмов исправления повреждений ДНК. Одна из таких систем - пострепликативная репарация, которая впервые была обнаружена у E. coli. Пострепликативная репарация происходит тогда, когда в ДНК возникает так много повреждений, что в ходе эксцизионной репарации клетка не успевает их полностью устранять, а также если повреждены гены, контролирующие синтез ферментов, участвующих в эксцизионной репарации.
Во время репликации ДНК с повреждением или ошибкой (например, пиримидиновым димером), ДНК-полимераза «перескакивает» через это повреждение и на вновь синтезируемой цепи остается брешь. Для устранения бреши происходит рекомбинация с участием особого белка RecA, - так, что в пустой промежуток поврежденной цепи встраивается комплементарный участок из неповрежденной «донорской» цепи, брешь на которой застраивается в процессе репаративного синтеза в процессе репликации. Пострепликативную репарацию называют также репарацией гомологичной рекомбинации.
Репарация разрывов двойной спирали у млекопитающих
До сих пор речь шла о репарации повреждений в пределах одной цепи ДНК. Рассмотрим, как происходит репарация обеих цепей ДНК при повреждении двойной спирали, например, ионизирующей радиацией. Как и при пострепликативной репарации, при репарации двуцепочечных разрывов происходит гомологичная рекомбинация, поскольку поврежденный участок ДНК замещается неповрежденным гомологичным участком. При разрыве сразу двух цепей в молекуле ДНК не остается неповрежденного гомологичного участка, поэтому ферменты «заимствуют» его из гомологичного участка сестринской хроматиды, и он встраивается на место поврежденного участка. Точный механизм такой репарации пока не ясен, но он позволяет сохранить непрерывность и целостность молекулы ДНК. Дефектность системы репарации двуцепочечных разрывов приводит к гиперчувствительности клеток к рентгеновским лучам, а также усиливает наследственную предрасположенность к раку груди и яичников.
Репарация ошибок репликации
Выше рассмотрены механизмы восстановления структуры ДНК после возникновения в ее молекулах различных повреждений. Однако при синтезе ДНК могут возникать просто ошибки репликации, которые делают возможным возникновение спонтанных мутаций. Несмотря на корректорские функции, присущие ДНК-полимеразам, некоторые нуклеотиды оказываются все же ошибочно включенными в новообразованную цепь ДНК. Для исправления ошибок репликации ДНК существуют другие системы репарации. Система исправления ошибок в ДНК также включает несколько различных ферментативных реакций. Процесс исправления начинается с распознавания некомплементарного нуклеотида и вырезания из цепи ДНК. За этим следует заполнение образовавшейся бреши в ходе репарационного синтеза с использованием в качестве матрицы верной нуклеотидной последовательности родительской цепи.
Заключая рассмотрение репарации ДНК необходимо отметить, что постоянно появляются все новые данные, свидетельствующие о большом разнообразии систем репарации ДНК, функционирующих как в прокариотических, так и эукариотических клетках. Эволюция этих систем несомненно обусловлена особым значением сохранности и точности передачи наследственной информации.
В сохранении стабильности функционирования генетического материала, кроме репарации ДНК, определенный вклад вносит вырожденность генетического кода. В силу вырожденности генетического кода мутация в виде замены нуклеотида может дать кодон-синоним, так что нормальная последовательность аминокислот в соответствующем полипептиде не изменится.
Понятие о генных болезнях
Говоря о сохранности и точности передачи наследственной информации надо признать, что никакая система передачи информации и никакие механизмы репарации ДНК не являются абсолютно совершенными и допускают с определенной частотой возникновение генных мутаций. Эти мутации проявляются фенотипически и, что важно для человека, проявляются в наследственные болезни. Наследственные болезни, обусловленные мутациями на генном уровне, получили название генные болезни. У человека отмечены следующие виды генных мутаций, обусловливающие генные болезни: миссенс, нонсенс, сдвиг рамки считывания, делеция, вставка (инсерция), нарушения сплайсинга, увеличение числа (экспансия) тринуклеотидных повторов. Число генных болезней у человека достаточно велико. В настоящее время описано более 5000 моногенных болезней человека. Ежегодно описываются новые моногенные болезни.
Начало развития любой генной болезни связана с первичным эффектом мутантного аллеля, поэтому принципиальные звенья развития (патогенеза) генных болезней можно представить следующим образом: мутантный аллель → патологический первичный продукт - белок (качественно или количественно измененный) → цепь последующих биохимических процессов → клетки → органы → организм.
Таким образом, в зависимости от контролируемого конкретным геном продукта и от характера нарушения его при мутации сначала развертывается развитие болезни на молекулярном уровне. Далее в определенных типах клеток разыгрываются основные патологические процессы, характерные для конкретного заболевания. Нарушения на клеточном уровне приводят к изменению состояния органов. Причем, при разных генных болезнях мишенью патологического процесса служат различные органы, иногда в результате первичных процессов, иногда - вторичных. В результате всего этого патологический процесс, защищенный первичным эффектом мутантного аллеля, приобретает целостность на организменном уровне с характерной клинической картиной. Ниже представлены некоторые моногенные болезни человека.
Особую группу моногенных наследственных болезней составляют болезни обмена веществ. Наследственные болезни обмена веществ подразделяют по типам обмена (углеводный, аминокислотный, обмен витаминов, липидов и др.). При этих заболеваниях четко выделяется первичный биохимический дефект. Заметное место среди генных наследственных болезней обмена веществ человека принадлежит заболеваниям, развивающимся вследствие мутаций генов, контролирующих образование белков-ферментов. Сюда, в частности, относятся фенилкетонурия, галактоземия, альбинизм и др.
Фенилкетонурия
Врожденное заболевание, вызванное нарушением перехода в организме фенилаланина в тирозин в силу рецессивной мутации гена фермента фенилаланингидроксилазы. В такой ситуации аминокислота фенилаланин не превращается в аминокислоту тирозин и накапливается в организме. Избыток фенилаланина и некоторых продуктов его обмена (фенилпировиноградной кислоты) оказывают токсическое действие на мозг ребенка, что приводит к задержке умственного развития. Частота встречаемости фенилкетонурии примерно 1:10000. Фенилкетонурия может быть также следствием нарушения сплайсинга. При этом нарушается сплайсинг 12 и 13 экзонов гена фенилаланингидроксилазы.
Галактоземия
Галактоземия - врожденное нарушение метаболизма галактозы, что приводит к накоплению ее в крови. Заболевание связано с рецессивной мутацией гена фермента галактозо-1-фосфат-уридилтрансферазы, превращающего галактозу в глюкозу. Повышенная концентрация в крови галактозы токсически влияет на организм, что проявляется увеличением печени, развитием катаракты, отставанием в умственном развитии. Характерны у новорожденного рвота и желтуха. Популяционная частота 3-5 на 1 млн.
Альбинизм
Врожденное заболевание, проявляющиеся отсутствием пигмента меланина в тканях. Меланин отсутствует в коже, волосах, радужной оболочки. Альбинизм обусловлен рецессивной мутацией гена белка-фермента тирозиназы, превращающий аминокислоту тирозин в меланин. Частота встречаемости 1:25000.
Муковисцидоз
Муковисцидоз - тяжелое наследственное заболевание с распространенным поражением дыхательных путей, поджелудочной железы и желез кишечника. Причиной заболевания в большинстве случаев является рецессивная мутация гена (делеция трех оснований), кодирующего трансмембранный регуляторный белок, обеспечивающий транспорт ионов хлора через мембраны эпителиальных клеток (служит каналом для ионов хлора). В силу мутации гена продуцируется белок с дефектом (отсутствие фенилаланина в 508 положении цепи белка), нарушающим выведение хлора из клетки. Нарушение транспорта хлора ведет к нарушению выделения воды из клетки в межклеточное пространство. Это ведет в свою очередь к дегидратации секрета как бронхов, так и протоков поджелудочной железы. Вязкий секрет закупоривает выводные протоки желез и бронхи, что приводит к воспалительному процессу с присоединением вторичной инфекции. Частота муковисцидоза составляет в среднем 1:2500 новорожденных.
Гемоглобинопатии
Гемоглобинопатии - это заболевания, связанные с наследственными нарушениями функции гемоглобина, обусловленные изменением в структуре его генов. Функционально полноценный гемоглобин представляет собой тетрамер. У взрослого человека он представлен четырьмя полипептидными глобиновыми цепями (двумя α-цепями и двумя β-цепями), соединенными с железосодержащим элементом - гемом. Полипептид-α включает 141 аминокислотный остаток, β-цепь - 146 аминокислотных остатков. Гены, контролирующие синтез обоих полипептидных цепей, расположены в разных хромосомах. Ген α-пептида расположен на коротком плече хромосомы 16, а β-пептида - на коротком плече хромосомы 11. У человека имеют место многочисленные мутации генов глобинов. Из них большинство редки (обычно не более 1% мутантных аллелей в аллелофонде популяции) и лишь некоторые из них встречаются относительно часто. Большая часть вариантов гемоглобина различается единичными аминокислотными заменами, причиной которых являются генные мутации, связанные с заменой нуклеотида в нуклеотидных последовательностях α- или β-глобиновых генов с изменением кода триплета. Если такие мутации затрагивают функционально значимые участки глобиновой полипептидной цепи: близкие к месту прикрепления гема, к местам контакта цепей друг с другом, влияющие на вторичную и третичную структуру молекулы - они, как правило, проявляются фенотипически в наследственные дефекты.
Широко известной формой аномальных гемоглобинов является серповидноклеточная анемия, при которой в 6-м положении β-цепи глютаминовая кислота замещена валином. При этом вместо нормального гемоглобина А будет синтезироваться гемоглобин S. Эта замена обуславливает пониженную растворимость гемоглобина, его повышенную полимеризацию. Гетерозиготные носители HbA/HbS в обычных условиях клинически здоровы, так как в крови у них содержится и нормальный гемоглобин (HbA). У гомозигот HbS/HbS с ранних лет развивается характерная картина хронической гипоксии и анемии с расстройствами кровообращения и тромбозами. В основе этих отклонений лежат полимеризация гемоглобина, изменение формы эритроцитов, их преждевременный гемолиз и распад.
Кроме замены отдельных нуклеотидов имеются и другие типы мутаций в генах гемоглобина (делеции, вставки, сдвиг рамки считывания), которые приводят к укорочению или удлинению полипептидов, или к прекращению их синтеза. Все они могут быть причиной развития наследственной патологии, объединеных в семейство гемоглобинопатий. Если результатом мутаций является подавление образования соответствующего полипептида гемоглобина (α или β), то это приводит к развитию патологических состояний, известных как талассемии. Такие состояния называют α- и β-талассемиями. У гомозигот по талассемии резко снижается образование гемоглобина (HbA), что вызывает гибель на стадии плода или в первые месяцы жизни ребенка.
Синдром Марфана
Это генетическое заболевание, возникающее в результате доминантной мутации гена (в основном миссенс), ответственного за синтез белка соединительной ткани фибриллина. Мутация ведет к нарушению синтеза фибриллина и развитию патологии соединительной ткани. Специфическими для диагностики синдрома Марфана являются нарушения в скелете, подвывих хрусталика глаза и пороки сердца. Частота синдрома Марфана составляет в среднем 1:10000 - 1:15000 новорожденных. Синдромом Марфана страдал президент США Авраам Линкольн, президент Франции Шарль де Голь и великий итальянский скрипач Николо Паганини.
К развитию наследственных заболеваний у человека приводит нарушение процессов репарации ДНК. Такие заболевания получили название - болезни репарации ДНК. К ним относятся: синдромы спонтанной хромосомной нестабильности (анемия Фанкони, синдром Блума, атаксия-телеангиэктазия), являющиеся редкими аутосомно-рецессивными заболеваниями; прогерия детей (синдром Хатчинсона-Гилфорда), прогерия взрослых (синдром Вернера). С нарушением репарирующих систем связано развитие редкого генетическкого заболевания человека - пигментной ксеродермии. У таких людей нарушена способность к эксцизионной репарации повреждений, вызванных ультрафиолетовым облучением, что связано с мутацией генов эксцизионной репарации. Солнечный свет, содержащий ультрафиолетовые лучи, вызывает у них веснушки и язвочки на коже, а также развитие рака кожи. Показано, что при эаболеваниях с наследственной предрасположенностью (подагра, шизофрения) также имеют место нарушения репарации ДНК.