

книги студ / Color Atlas of Pathophysiology (S Silbernagl et al, Thieme 2000)
.pdf
|
|
|
|
|
A. Disorders of Sleep Regulation |
|
|
|
B. Sleep Phases |
|
|
|
|
Cortex |
Somnambulism |
Awake |
|
|
|
|
|
Normal |
|
||||
|
|
|
|
|
|
|
|
|
|||||||
Lesions, |
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
sleep |
|
|
|
|
|
|
|
|
|
|
ischemia |
|
|
Asynchronous |
I |
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
of |
II |
|
|
|
|
|
|
|
|
|
|
Thalamus |
|
Somnolence |
Stages |
III |
|
|
|
|
|
REM |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
SCN |
Hypo- |
|
|
|
IV |
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
thalamus |
|
|
ARAS |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NR |
|
Awake |
|
Excessive use of sleeping pills |
Disorders |
|||||||
|
|
Reticular |
|
|
|
||||||||||
|
|
formation |
LC |
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|||||
Insomnia, hypersomnia |
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
I |
|
|
|
|
|
|
|
Sleep |
|||
Metabolic |
|
|
Respiratory |
|
II |
|
|
|
|
|
|
|
|||
alkalosis |
|
|
|
center |
|
III |
|
|
|
|
|
|
|
10.22 |
|
Abnormal |
Muscle tone |
|
|
|
IV |
|
|
|
|
|
|
|
|||
regulation |
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
Stridor |
|
|
|
|
|
|
|
|
|
|
|
||
|
|
Sleep apnea |
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
Plate |
|||
|
|
|
|
|
|||||||||||
|
|
|
|
|
Hypoxia |
|
|
|
|
Hours |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
C. Sleep Factors |
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
Normal sleep phases |
|
|
||||
Sleep pressure |
1 |
Day |
Night |
|
NREM sleep pressure |
|
|
|
|
|
|
|
|||
|
|
1/REM sleep pressure |
|
Net sleep pressure |
|
Depth of sleep |
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
‘Normal’ on- |
|
|
|
|
Tage |
|
|
|
|
|
||
|
|
|
set of night |
Jet lag |
|
|
Delayed sleep phase insomnia |
|
|
||||||
|
2 |
|
|
|
|
3 |
|
|
|||||||
|
East |
West |
|
|
|
|
Diurnal rhythm |
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
displaced |
|
|
|
|
|
|
|
|
|
|
|
Depression |
|
|
|
|
Excitement |
|
|
|||
|
4 |
|
|
|
Sleep |
|
5 |
|
Raised level |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
of excitement |
|
|
|
|
|
||
|
Serotonin deficiency (?) |
|
deprivation |
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
341 |
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.

10 Neuromuscular and Sensory Systems
342
Consciousness
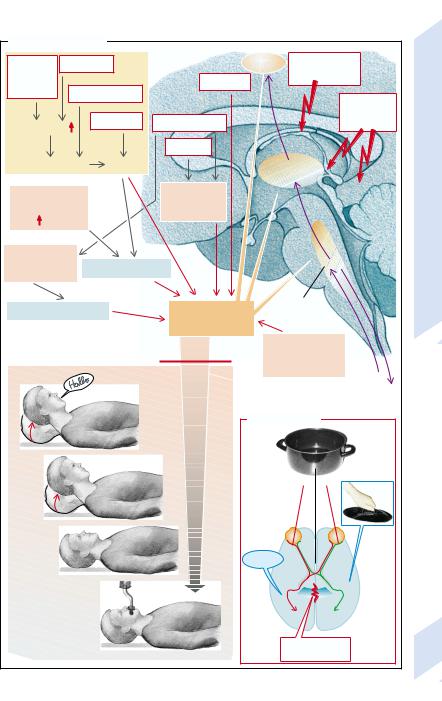
We become conscious of only a fraction of the information reaching our brain. The conscious contents are stored in associative cortical areas that specialize in this task (→p. 346). Conscious awareness requires not only that the specific afferents have been transmitted to the cerebral cortex, but also nonspecific activation by the ARAS through which neurons from the reticular formation activate wide areas of the cerebral cortex via intralaminar neurons of the thalamus (→A).
Damage to large areas of the cortex and/or breakdown of the ARAS brings about loss of consciousness. In addition, there may be primary causes influencing neuronal excitability in the above-mentioned neuronal structures. Ischemia (e.g., atherosclerotic vascular occlusion) or hypoxia (e.g., suffocation) (→A1) impair excitability directly or by cell swelling. Swelling of glial cells impairs, among other functions, their capacity to take up K+ and thus to keep down the concentration of extracellular K+. This has an indirect effect on neuronal excitability. Part of the effect of tumors, abscesses, or bleeding is also exerted via ischemia or hypoxia (→A1) in that they raise the cerebral pressure and thus impair cerebral perfusion by narrowing the blood vessels. Hypoglycemia also modifies excitability, partly via cellular swelling (→A2). Hyponatremia and ammonia (NH4+) also act via this mechanism. The rise in NH4+ in hepatic encephalopathy (→p.174) causes the formation of glutamine from α-ketoglutarate and glutamate in glial cells; the accumulation of glutamine causes them to swell. At first this swelling is counteracted by the removal of osmolytes, seen in magnetic resonance imaging as a decrease in the cerebral concentration of inositol. When this compensatory mechanism is exhausted, consciousness is lost.
The excitability of neurons is also affected by epilepsy (→p. 338), hyperosmolarity (hypernatremia, hyperglycemia; →A3) as well as by disorders of electrolyte (Ca2+, Mg2+, HPO42–) and acid-base metabolism (→A4). Uremia (in renal failure) and diabetes mellitus act partly via changes in extracellular osmolarity and electrolyte composition. Numerous substances can impair the excitability of the
ARAS (→A5), such as NMDA receptor antagonists, alcohol, narcotics, hypnotics, psychoactive drugs, anticonvulsives, Na+/K+-ATPase inhibitors (cardiac glycosides), heavy metals. Extreme excess or lack of hormones (e.g T3, T4, parathyroid hormone, adrenocorticoid hormones, pheochromocytoma) as well as massive neuronal excitation, for example, caused by pain or psychogenic disease (schizophrenia), can lead to loss of consciousness (→A6). Lastly, neuronal excitability can also be so severely impaired by hyperthyroidism, hypothermia, inflammatory (e.g., meningitis) or mechanical damage, and neurodegenerative disease that it could lead to loss of consciousness (→A7).
Loss of consciousness can be divided into several stages (→A): in a state of drowsiness the patient can still be roused and will respond; in a stupor (profound sleep) patients can be awakened by vigorous stimuli; when in a coma this is no longer possible. In socalled “coma dépassé” vital functions will also have ceased (e.g., respiratory arrest).
The split brain represents a special abnormality of consciousness (→B). Uniform consciousness presupposes communication between the two cerebral hemispheres. This takes place along large commissural fiber bundles through the corpus callosum and the anterior commissure. In treating uncontrollable epilepsy the commissural fibers have been transected in some patients, stopping this communication between the two hemispheres. The two hemispheres now produce two distinct kinds of consciousness: if an object (e.g., a saucepan) is placed into the right hand or placed in the right visual field, the patient can correctly name the object. But if the object is placed into the left hand or projected into the left visual field, the patient is able to recognize the object and, for example, find the appropriate saucepan cover with the left hand, but will not be able to name it.
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.
A. Unconsciousness |
|
|
|
|
|
|
|
||
Bleeding, |
Injury |
1 |
|
|
Cortex |
Excessive |
|
6 |
|
tumor, |
|
|
|
|
|
hormonal disorder |
|
||
abscess, |
|
|
Epilepsy |
|
|
||||
|
|
|
|
|
|
|
|||
cerebral |
|
|
|
|
|
|
|
||
edema |
Vascular occlusion |
|
|
|
|
|
Alcohol, |
5 |
|
|
|
|
|
|
|
|
|
||
|
Asphyxiation |
|
|
|
|
|
narcotics, |
||
Cerebral pressure |
Diabetes mellitus |
|
|
|
toxins |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
Uremia |
|
|
|
|
|
|
Ischemia |
Hypoxia |
|
|
Intralaminar |
|
|
|
||
|
|
|
|
|
thalamic nuclei |
|
|
Consciousness |
|
Hypoglycemia, |
2 |
|
Electrolyte |
3 |
|
|
|
|
|
|
disorders, |
|
|
|
|
|
|||
hyponatremia, |
|
|
+ |
|
|
|
|
||
|
|
abnormal H |
|
ARAS |
|
|
|||
NH4+ |
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|||
Hypernatremia,4 |
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
10.23 |
||
hyperglycemia |
|
Cell swelling |
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
Cell shrinkage |
|
Neuronal |
|
Reticular |
|
|
Plate |
||
|
|
formation |
|
|
|||||
|
excitability |
|
|
|
|
||||
|
|
|
|
|
|
|
|
||
|
|
|
in cortex |
|
|
|
|
|
|
|
|
|
Unconsciousness |
Temperature, |
7 |
|
|
||
|
|
|
inflammation, |
|
|
|
|||
|
|
|
|
|
neurodegeneration |
|
|
||
Somnolence |
|
|
|
|
|
|
|
Sensory |
|
|
|
|
|
|
|
|
|
system |
|
|
|
|
|
|
|
|
|
Motor |
|
|
|
|
|
|
|
|
|
system |
|
|
|
|
|
|
B. ‘Split brain’ |
|
|
|
|
Stupor |
|
|
|
|
|
|
|
„?“ |
|
Coma |
|
|
|
|
“Sauce- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
pan” |
|
|
|
|
‘coma dépassé’ |
|
|
|
Left |
|
|
Right |
||
|
|
|
|
|
hemisphere |
|
hemisphere |
||
|
|
|
|
|
|
Corpus callosum |
343 |
||
|
|
|
|
|
|
transected |
|
||
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.

10 Neuromuscular and Sensory Systems
344
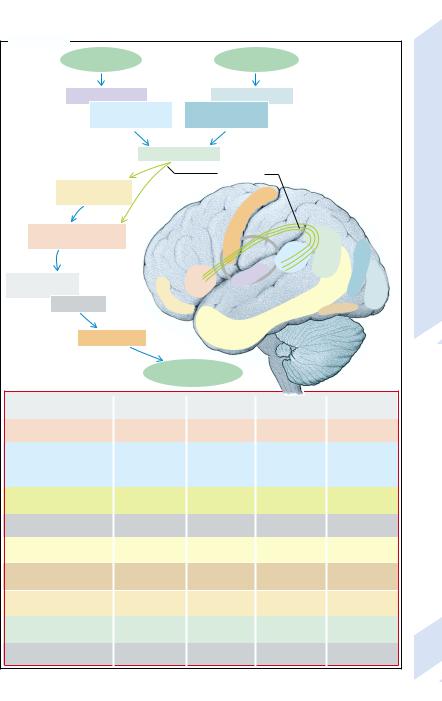
Aphasias
Speech and language comprehension are tasks |
of “spin”) or semantic errors (“mother” instead |
that engage a large part of the cerebral cortex. |
of “woman” [paraphasia]) or create new words |
For this reason, lesions in various parts of the |
(neologisms). |
cortex may lead to an impairment of speech |
In conductive aphasia the connection be- |
and of language comprehension. |
tween sensory and motor speech center (arcu- |
Simply put, spoken language is first per- |
ate fasciculus) is interrupted. Speech is fluent |
ceived in the primary auditory cortex (→A; |
(although sometimes paraphasic) and com- |
marked in violet) and then in the sensory |
prehension is good. However, their repetition |
speech center (Wernicke’s area, marked in |
ability is greatly impaired. They are also un- |
light blue). Written words are transmitted via |
able to read aloud, even though they under- |
the primary (gray-blue) and secondary (dark |
stand the text they read. |
blue) visual cortex to area 39, where acoustic, |
In global aphasia (damage to both the sen- |
optical, and sensory perceptions are integrat- |
sory and the motor centers, e.g., by occlusion |
ed. When writing, the premotor cortex is |
of the medial cerebral artery) both sponta- |
activated via the arcuate fasciculus of the pre- |
neous speech and comprehension are im- |
motor cortex that, in turn, activates the motor |
paired. |
cortex via the basal ganglia and the thalamus. |
Anomic aphasia is the result of a lesion in |
In right-handed people the structures involved |
the temporal lobe in the region of the medial |
are predominantly localized in the left hemi- |
and inferior gyri. Patients’ speech is largely |
sphere, and speech disorders (aphasia) are al- |
normal but it is difficult for them to find the |
most always the result of lesions in the left |
right word for certain objects. In achromatic |
hemisphere. |
aphasia (lesion at the left inferior temporal |
Each of the above-mentioned structures can |
lobe close to temporal-occipital border) the |
cease functioning, for example, due to trau- |
person cannot name a color (even though it is |
matic or ischemic damage. Depending on |
correctly recognized and objects can normally |
which cerebral area is affected, abnormalities |
be sorted by color). |
characteristic for each will develop. |
Transcortical motor aphasia is caused by a |
Broca’s aphasia is caused by a lesion of the |
lesion in the anterior inferior frontal lobe near |
motor speech center in area 44 and the neigh- |
the Broca speech center. Spontaneous speech |
boring areas 9, 46, and 47. Spontaneous speech |
is markedly impaired, while repetition and |
(verbal output) is grammatically incorrect and |
comprehension are normal. |
the patient typically communicates by using |
Transcortical sensory aphasia occurs after a |
single words and is incapable of repeating |
lesion in the parietal–temporal association |
someone else’s words (impaired repitition |
cortex near the Wernicke speech center or |
ability). Language comphrehension is not, or |
area 39. The patient can speak fluently and |
less markedly, impaired. As a rule patients |
repetition is normal. However, there is a prob- |
cannot write normally. However, if the lesion |
lem understanding words and finding the |
is limited to area 44, the ability to write is pre- |
right word; reading and writing are impossi- |
served (a rare disorder, called aphemia). |
ble. |
Wernicke’s aphasia results from a lesion in |
Subcortical aphasia is due to lesions in the |
the sensory speech region, i.e., in the posterior |
region of the basal ganglia (especially the cau- |
portion of the temporal gyrus of the auditory |
date nucleus) and the thalamus. There are |
association cortex (area 22) and/or the supra- |
transient disorders of comprehension and |
marginal gyrus (area 40). Language compre- |
finding of words. |
hension is impaired in these patients. At the |
|
same time they also lose the ability to repeat |
|
words spoken by somebody else. Spontaneous |
|
speech is fluent; sometimes patients speak all |
|
the time (logorrhea). However, in doing so they |
|
may make occasional phonetic (“spill” instead |
|
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.
A. Aphasias |
|
|
|
|
|
|
Word |
Word |
|
|
|
||
which is heard |
which is read |
|
|
|
||
Primary auditory cortex |
Primary visual cortex |
|
|
|
||
|
Secondary |
Secondary |
|
|
|
|
auditory cortex |
|
|
|
|
||
visual cortex |
|
|
|
|
||
(Wernicke’s area) |
|
|
|
|
||
|
|
|
|
|
||
|
Area 39 |
|
|
|
|
|
|
|
Arcuate |
|
|
|
|
|
|
fasciculus |
|
|
|
|
Anterior superior |
|
|
|
|
|
|
frontal lobe |
|
Motor |
|
|
|
|
|
|
cortex |
|
|
|
Aphasias |
Premotor cortex |
|
|
|
|
|
|
(Broca’s area) |
|
|
|
|
Secondary |
|
|
|
|
|
Area 39 |
visual cortex |
10.24 |
|
|
|
|
|
||
Basal ganglia, |
Broca’s |
Wernicke’s |
|
Primary |
||
area |
Primary |
area |
|
visual |
||
cerebellum |
Anterior |
auditory |
|
|
cortex |
Plate |
Thalamus |
inferior |
cortex |
|
|
|
|
frontal lobe |
|
|
|
|||
|
|
|
|
|||
|
|
|
|
|
|
|
Motor cortex |
|
|
|
|
|
|
|
Spoken word |
|
|
|
|
|
Type |
Spontaneous |
Repetition of |
Language |
Finding words |
|
|
|
speech |
words |
comprehension |
|
|
|
Broca’s aphasia |
abnormal |
abnormal |
normal |
|
impaired |
|
Wernicke’s aphasia |
fluent |
abnormal |
impaired |
|
impaired |
|
|
(at times logorrhea, |
|
|
|
|
|
|
paraphasia, |
|
|
|
|
|
|
neologisms) |
|
|
|
|
|
Conduction aphasia |
fluent, |
markedly |
normal |
|
abnormal, |
|
|
but paraphasic |
impaired |
|
|
paraphasic |
|
Global aphasia |
abnormal |
abnormal |
abnormal |
|
abnormal |
|
Anomic aphasia |
fluent |
normal, |
normal |
|
impaired |
|
|
|
but anomic |
|
|
|
|
Achromatic aphasia |
fluent |
normal, |
normal |
|
impaired |
|
|
|
but anomic |
|
|
|
|
Motor transcortical |
abnormal |
normal |
normal |
|
abnormal |
|
aphasia |
|
|
|
|
|
|
Sensory transcortical |
fluent |
fluent |
abnormal |
|
abnormal |
|
aphasia |
|
|
|
|
|
|
Subcortical aphasia |
fluent |
normal |
abnormal |
|
abnormal |
345 |
|
|
|
(transient) |
|
(transient) |
|
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.

10 Neuromuscular and Sensory Systems
346
Disorders of Memory
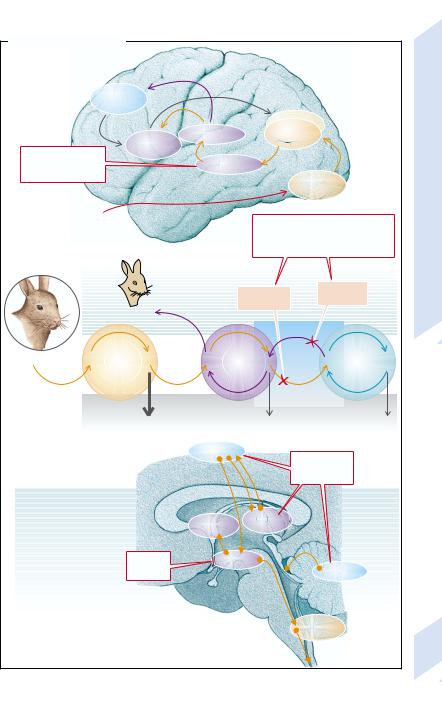
Two forms of memory are distinguished: Declarative, explicit memory (semantic or episodic) stores memory that can only be recalled consciously (→A). It is needed, for example, in order to be able to recognize certain things (apples, animals, faces). Procedural, implicit memory (→A3) does not require conscious activation for storage and recall. It is required, e.g. for learning to play the piano.
To form declarative memory (→A1) the information first of all reaches the corresponding association cortex (e.g., the secondary visual cortex) via the particular primary sensory cortical area (e.g., the primary visual cortex). From here, via the entorhinal cortex (area 28), the information reaches the hippocampus, which is essential for long-term storage of declarative memory. With mediation from structures in the diencephalon, basal forebrain, and prefrontal cortex the item is again stored in the asssociation cortex. In this way the information is first taken up, via the sensory memory, by the short-term memory, which can hold on to the content for only a few seconds to minutes. The information can be transferred to the long-term memory, for example, through being rehearsed (→A2). Such rehearsal is not an essential precondition for the formation of long-term memory, however.
It is particularly the transfer into long-term memory that is impaired in lesions of the above-named structures in neurodegenerative diseases (e.g., Alzheimer’s disease; →p. 348), trauma, ischemia, alcohol, carbon monoxide, and inflammation. In addition, memory formation can be temporarily stopped by electric shock. The most important transmitter in the hippocampus is glutamate (NMDA receptors). Memory formation is promoted by norepinephrine and acetylcholine (nicotinergic receptors).
Lesions in the hippocampus or its connections result in anterograde amnesia (→A2). The affected patients will from that moment on no longer be able to form any new declarative memory. They will remember events prior to the lesion but none subsequent to it.
Retrograde amnesia (→A2), i.e., the loss of already stored information, occurs in disorders in the relevant associative cortical fields. De-
pending on the extent and localization of the disorder, the loss can be reversible or irreversible. In the former case the patient will lose items of memory, but they can be retrieved. In irreversible loss the particular items are permanently lost.
Transitory bilateral functional disturbance of the hippocampus can cause anterograde and retrograde (days to years) amnesia (transient global amnesia). In Korsakoff’s syndrome
(frequent in chronic alcoholics) both anterograde and retrograde amnesia can occur. Patients thus affected often try to cover up gaps in memory by means of confabulations.
The procedural (implicit) memory (→A3) is not impaired in lesions of the hippocampus. It allows imprinting, learning of skills, sensitization, habituation, and conditioning. Depending on the task, cerebellum, basal ganglia, amygdala and cortical areas are involved. Both the cerebellum and basal ganglia play an important role when learning skills. Relevant afferent impulses reach the cerebellum via olivary and pontine nuclei. The storage capacity of the cerebellum can be lost by, for example, toxic damage, degenerative diseases, and trauma. Dopaminergic projections of the substantia nigra also play a part in the formation of procedural memory.
The amygdala is important in conditioning anxiety reactions. It receives its information from the cortex and thalamus and influences motor and autonomic functions (e.g., muscle tone, palpitations [awareness of tachycardias], goose-pimples) via the reticular formation and hypothalamus. Removal of the amygdala (e.g., by trauma or opiates) cancels conditioned anxiety reactions. Bilateral removal of the amygdala with portions of the hippocampus and temporal lobe results in amnesia and disinhibited behavior (Klüver–Bucy syndrome).
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.
A. Disorders of Memory |
|
|
|
|
|
|
|
Prefrontal |
|
|
|
|
|
|
|
|
cortex |
|
|
|
|
|
|
|
Basal |
Diencephalon |
|
|
Association |
|
|
|
|
|
cortex |
|
|
||
|
prosencephalon |
|
|
|
|
||
Glutamate (NMDA), |
|
|
|
|
Memory |
||
|
|
|
|
|
|
||
acetylcholine, nicotine, |
|
Hippocampus |
|
|
|||
norepinephrine |
|
|
|
|
Sensory |
||
|
|
|
|
|
|||
|
|
|
|
|
cortex |
||
|
|
|
|
|
of |
||
|
|
|
|
|
|
|
|
Sensory |
|
|
|
|
|
|
Disorders |
perception |
|
|
|
|
Trauma, |
Degeneration, |
|
1 |
|
|
|
|
|||
|
|
|
|
tumors, |
alcohol, CO, |
||
Declarative memory |
|
|
|
|
|||
|
|
|
|
inflammation, |
electric shock, |
||
|
|
|
|
|
ischemia |
epilepsy |
|
|
|
|
|
|
|
||
|
|
Remembering |
|
|
|
Retrograde |
10.25 |
|
|
Anterograde |
Plate |
||||
|
|
|
amnesia |
||||
|
|
|
|
amnesia |
|||
|
‘Rabbit’ |
|
|
|
|||
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
Perception |
Sensory |
Short-term |
Re- |
Long-term |
|
||
hearsal |
|
||||||
|
memory |
memory |
|
memory |
|
||
|
|
|
|
||||
|
Second |
Minutes |
|
|
Years |
|
|
2 |
|
Forgetting |
|
|
|
|
|
|
|
|
|
|
|
|
|
Model of cognitive learning |
|
|
|
|
|
|
|
|
|
Cortex |
|
|
Toxins, |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
degeneration, |
|
|
|
|
|
|
|
trauma |
|
|
Imprinting, |
|
Hypo- |
|
Basal |
|
|
|
skills |
|
|
ganglia |
|
|
||
|
thalamus |
|
|
|
|||
sensitization, |
|
|
|
|
|
|
|
habituation, |
Trauma, |
Amygdala |
|
|
|
||
conditioning |
Cerebellum |
|
|||||
|
opiates |
|
|
|
|
||
3 |
|
|
|
|
Reticular |
|
|
|
|
|
|
formation |
|
||
Procedural learning |
|
|
|
||||
|
|
|
|
|
|||
|
|
|
|
|
|
|
347 |
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.

10 Neuromuscular and Sensory Systems
348
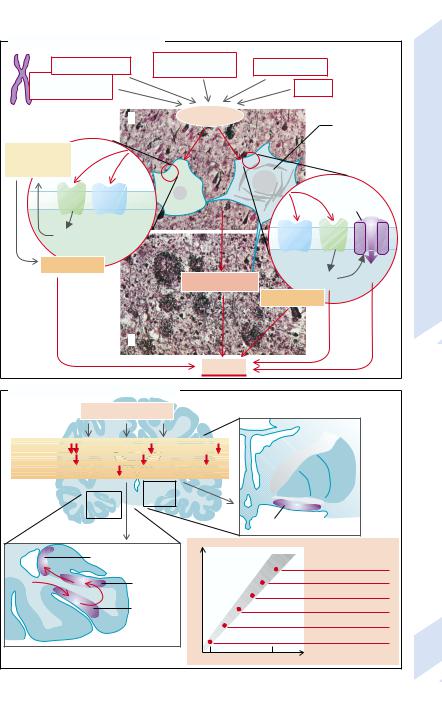
Alzheimer’s Disease
The occurrence of Alzheimer’s disease, the most common cause of (senile) dementia (about 70%), is favored by a genetic disposition. However, the disease is not genetically uniform. An especially severe form of the disease has an autosomal dominant inheritance. Defects on chromosomes 1, 12, 14, 19, or 21 were found in families with Alzheimer’s disease. The defective gene on chromosome 19, for example, codes for apolipoprotein E (ApoE 4), the relevant gene on chromosome 21 for a protein (β-amyloid precursor) that can be broken down to small amyloid peptides. These can on their own bunch themselves together into protein fibrils 7 – 10 nm long (→A1). These amyloid fibrils can then form aggregates, 10 µm to several hundred µm in diameter (senile plaques), that are frequently found in the brain of patients with Alzheimer’s disease (→A2). In addition to extracellular amyloid, these plaques contain distorted dendrites and axons with abnormal intracellular neurofibrils. The formation of these atypical elements of the cytoskeleton apparently precedes the death of the neurons (see below).
Certain mutations of the β-amyloid precursor gene promote the formation of senile plaques. Amyloid deposits can also occur under the influence of other genetic or external factors. It is thought, for example, that toxins can penetrate the brain via the olfactory nerves and cause the disease. Amyloid deposits also occur in trisomy 21 (Down’s syndrome) that also leads to dementia.
β-amyloid fibrils can react with receptors at the cell surface, such as the receptor for advanced glycation end products (RAGE), and a scavenger receptor (RA). Oxygen radicals formed as a result may increase the neuronal intracellular concentration of Ca2+ (→A3), possibly via depolarization of the cell membrane and activation of NMDA receptors. The O2 radicals and Ca2+ promote cell death. In microglial cells (→A4) the activation of RAGE and RA stimulates the formation or release, respectively, of NO, prostaglandins, excitotoxins, cytokines, tumor necrosis factor (TNF-α), tumor growth factor (TGF-β1), and fibroblast growth factor (b-FGF). This results in inflammation that also impairs neurons. Increased concen-
tration of the osmolyte inositol points to a disorder of cell volume regulation.
The death of neurons is accelerated by a lack of NGF or of NGF receptors and can be delayed by NGF.
Cholinergic neurons in the basal nucleus of Meynert, in the hippocampus (especially CA1, the subiculum) and in the entorhinal cortex (→B1) are particularly affected by cell death, but neurons also die in other cerebral areas, such as the frontal lobes, anterior temporal lobes, parietal lobes, olfactory cortex, hypothalamus, locus ceruleus, and raphe nuclei.
Neuronal death is accompanied by decreased formation and concentration of neurotransmitters in the brain. Acetylcholine is markedly affected: in the cerebral cortex and the hippocampus there is an up to 90% decrease in the concentration of choline-acetyl transferase, the enzyme that is necessary for the formation of acetylcholine. The concentration of other neurotransmitters is also reduced, for example, norepinephrine, serotonin, somatotropin, neuropeptide Y, substance P, and corticotropin-releasing hormone ([CRH] corticoliberin).
A consequence of the degenerative changes is an increased loss of cerebral functions
(→B2). The disease typically begins insidiously with subtle deficits of memory, neglect of appearance and body hygiene, phases of confusion, and taking wrong decisions. As the disease progresses, anterograde amnesia (→ p. 346) will be followed by impairment of past memories as well as procedural memory. Lesions in the limbic system express themselves alternately through restlessness and lethargy. Motor deficits (speech disorders, abnormal muscle tone, ataxia, hyperkinesia, myoclonus) occur relatively late.
Creutzfeldt–Jakob disease, possibly caused by prions (proteinaceous infectious particles), is a neurodegenerative disease that, in addition to motor (e.g., ataxia) and psychogenic disorders, also leads to dementia.
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.
A. Causes of Alzheimer’s Disease |
|
|
|
|
||
Normal protein |
Other genetic factors |
Environmental |
|
|
|
|
Defective β-amyloid |
|
factors |
|
|
|
|
|
Toxins |
|
|
|
||
precursor |
|
|
|
|
|
|
|
|
|
|
|
|
|
Long arm of |
1 |
Amyloid |
|
|
|
|
chromosome 21 |
|
|
Abnormal |
|
||
|
|
|
|
|
||
|
|
|
|
|
neurofibrils |
|
NO, exitotoxins, |
|
|
|
|
|
Disease |
TNFα, TGFβ, |
|
|
|
|
|
|
bFGF |
|
|
|
|
|
|
RA |
RAGE |
|
|
|
NMDA |
|
|
|
|
Alzheimer’s |
|||
|
|
|
|
|
receptor |
|
O2– |
4 |
|
RAGE |
RA |
||
Glial cell |
|
|||||
|
|
|
|
|
||
|
|
|
|
|
|
|
Inflammation |
|
3 |
|
Ca2+ |
10.26 |
|
|
|
Senile plaques |
O2– |
|||
|
|
|
NGF deficiency |
|
Neuron |
Plate |
|
|
|
|
|
||
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
Cell death |
|
|
|
|
Photos: Doerr W. et al.. Atlas der pathologischen Anatomie. Stuttgart: Thieme; 1975.
B. Effects of Alzheimer’s Disease
|
Neuronal death |
|
|
Acetylcholine |
Neuropeptides |
Norepinephrine |
|
Somatostatin |
Substance P |
Serotonin |
Putamen |
|
CRF |
|
Globus |
|
|
|
|
|
|
|
pallidus |
|
|
1 |
Basal nucleus of Meynert |
CA1 |
Hippocampus |
|
|
Complete loss of |
|
|
|
|
|
||
|
|
|
mental control |
|
|
|
|
|
|
|
|
|
Subiculum |
defects |
|
Motor seizures |
|
|
|
Global amnesia |
|
||
|
Entorhinal |
|
|
||
|
|
|
|
Lethargy |
|
|
cortex |
Mental |
|
Anterograde amnesia |
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
Forgetfulness |
|
|
|
60 |
70 Years |
2 |
349 |
|
|
|
|
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.

10 Neuromuscular and Sensory Systems
350
Depression
Depression is a disease with an increased familial incidence. It can alternate with manic phases (bipolar disorder) or can occur in isolation (unipolar disorder).
Pathophysiologically, depression is thought to be connected with decreased (relative or absolute) availability of norepinephrine and/or serotonin in the brain.
Norepinephrine is formed in neurons of the locus ceruleus and the tegmentum (→A). Axons from the tegmentum predominantly connect with the hypothalamus, anterior pituitary, brain stem, and spinal cord. Fibers from the locus ceruleus project to spinal cord, hypothalamus, thalamus, limbic system, and cortex.
The release and action of norepinephrine at the nerve endings can be reduced by a number of substances, leading to depression (→A1):
The synthesis of norepinephrine from tyrosine via DOPA can be reduced by enzyme inhibitors (e.g., methyltyrosine).
The uptake of norepinephrine in presynaptic
stores can be inhibited (e.g., by reserpine).
Norepinephrine can be replaced at the postsynaptic receptors (e.g., phenoxybenzamine, phentolamine).
The synaptic norepinephrine concentration and action can, however, also be increased, an effect which is in part utilized in the drug treatment of depression (→A2).
Inhibitors of monoamine oxidase A (MAO- A), which is specific to norepinephrine (and serotonin) (e.g., tranylcypromine, moclobemide), can delay the breakdown of norepinephrine in the presynaptic endings and thus increase its availability.
Inhibitory substances of catechol-ortho- methyl-transferase ([COMT] e.g., tropolone) delay the breakdown of norepinephrine.
Amphetamines increase synaptic concentration of norepinephrine, dopamine, and serotonin by inhibiting transport of the transmitter.
Desipramine inhibits re-uptake, and thus similarly increases the synaptic norepinephrine concentration.
The receptors can be stimulated by agonists (e.g., clonidine).
Serotonin (5-hydroxytryptamine [5-HT]) is formed in neurons of the raphe nuclei that project to the spinal cord, cerebellum, thalamus, hypothalamus, basal ganglia, the limbic system, and cerebral cortex (→B).
A reduced availability or action of serotonin (→B1) favors development of depression, for example
by inhibiting synthesis from tryptophan (e.g., chlorophenylalanine);
by inhibiting uptake in presynaptic stores (e.g., reserpine);
due to increased consumption of serotonin through formation of inactive melatonin (when dark, in the pineal gland).
An antidepressive effect has been observed when serotonin action or stimulation of the serotonin receptors has been increased (→
B2):
Availability of tryptophan can be increased by administering glucose. Glucose promotes insulin release, and the antiproteolytic and protein synthesis-stimulating effect of insulin leads to a reduction of amino acid concentration in blood. Some amino acids competitively inhibit tryptophan uptake across the blood– brain barrier. Loss of this inhibition would raise tryptophan uptake in the brain.
Tricyclic antidepressants (e.g., imipramine, fluoxetine) inhibit the re-uptake of serotonin in presynaptic stores and in this manner also increase its synaptic concentration.
MAO-A inhibitors (see above) raise the availability of serotonin by inhibiting its breakdown.
Exposure to light inhibits the conversion of serotonin to melatonin. Because of the short and relatively dark days, depression is particularly frequent in northern countries during the winter months. Conversely, depression can sometimes be succesfully treated by exposing patients to bright light (phototherapy).
Agonists (e.g., lysergic acid diethylamide [LSD]) can directly stimulate serotonin receptors.
Lithium probably exerts its antidepressive effect by influencing intracellular signal transmission (→p. 6).
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.