

книги студ / Color Atlas of Pathophysiology (S Silbernagl et al, Thieme 2000)
.pdfA. Acute Primary Gout |
|
|
|
|
|
|
|
AMP |
APRT |
Adenine |
|
Nucleotide |
|
||
|
|
|
|
|
breakdown |
|
|
IMP |
Inosine |
|
|
Hypoxanthine |
|
|
|
|
|
|
|
|
XO |
Allopurinol |
10% of |
|
HGPRT |
|
|
filtered urate |
|||
|
|
|
|
|
|||
GMP |
Guanosine |
|
Guanine |
|
Xanthine |
Benz- |
|
|
|
bromarone |
|||||
|
|
|
|
|
|
|
|
|
|
O |
|
|
|
XO |
|
1 |
|
|
|
|
|
2 |
|
|
|
|
H |
|
|
||
|
|
C |
|
Uric acid formation |
Uric acid excretion |
||
|
HN |
C |
N |
||||
|
|
|
|
|
|||
|
|
|
|
|
|
||
|
|
|
|
C |
O |
|
|
|
O |
N |
C |
N |
|
|
|
|
C |
|
|
|
|
||
|
|
H |
|
H |
|
|
|
High purine intake with food
Rare: partial
HGPRT defects
|
|
|
|
|
|
|
Asymptomatic hyperuricemia |
|
|
|
|
|
|
|
|
|
Yearsto decades |
Alcohol |
|
|
|
Cold |
|
|
|
|
|||||
|
|
|
|
|
|
||
|
|
Thiazides |
|||||
3 |
|
|
|
|
|
|
Obesity |
|
Lead pollution |
||||||
Precipitation of sodium urate crystals in joints (microtophi)
|
|
Trauma? |
|
|
|
|
|
|
|
|
Sudden rise |
|
Release of sodium urate crystals: attack of gout |
in plasma urate |
|||
|
High urine osmolality |
||||
|
Complement activation and chemotaxis |
|
|||
4 |
|
|
|||
|
of: |
|
|
Low urine pH |
|
|
|
|
|
||
|
Monocytes/macrophages |
Neutrophils |
|
|
|
IL-1, -6, -8; TNFα |
Phagocytosis |
|
5 |
||
|
|
|
of crystals |
|
|
Cell damage |
|
Cell |
Release |
Urate precipitation |
|
|
of crystals |
||||
|
|
Leukotriene B4, |
death |
in urine |
|
|
|
|
|||
|
|
|
|
|
|
|
|
prostaglandins, |
|
|
|
Protease |
O2 radicals |
Lysosomal |
|
Urate stones |
|
release |
|
enzymes |
|
||
|
|
Joints: |
|
|
Acute urate nephropathy |
|
acute inflammation, tissue damage |
|
|||
|
|
|
|||
|
|
|
Photo: Siegenthaler W. Differentialdiagnose innerer Krankheiten.16th ed. Stuttgart: Thieme: 1988. |
||
Plate 8.5 Gout
251
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.

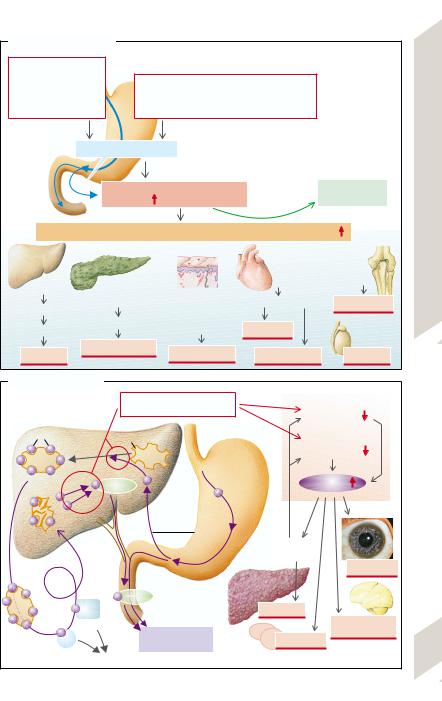
Hemochromatosis
|
Hemachromatosis is a condition in which |
||
|
there is an excessive accumulation of iron (Fe) |
||
|
in the body, deposited in the parenchymal cells |
||
|
of the liver, pancreas, and other organs. Pri- |
||
|
mary |
(= idiopathic, = hereditary) |
hemochro- |
|
matosis (→A1) is a common disease (1 in |
||
|
400), inherited as an autosomal recessive trait. |
||
|
The abnormality consists of a markedly raised |
||
|
intestinal absorption of Fe; in a year 0.5 –1 g ex- |
||
|
cess Fe is taken up. Fe, ferritin, and transferrin |
||
|
saturation are increased in serum (→p. 38). If |
||
|
diagnosed early, the increased Fe content |
||
|
(25 – 50 g) can be normalized by |
means of |
|
|
weekly bloodlettings over one to two years (se- |
||
Metabolism |
rum ferritin < 50 µg/L; transferrin saturation |
||
< 50%). Secondary hemochromatoses (→A2) |
|||
or sideroblastic anemia; →p. 36), liver disease |
|||
|
occur if there is an abnormal utilization of Fe |
||
|
(increased Fe absorption with ineffective |
||
|
erythropoiesis, for example, in β thalassemia |
||
8 |
(e.g., alcoholic cirrhosis, portocaval shunt), in |
||
|
atransferrinemia (→p. 38), and porphyria cu- |
||
|
tanea tarda (→p. 254) as well as in excessive |
||
|
Fe supply, either orally or parenterally (fre- |
||
|
quent blood transfusions which are a second |
||
|
cause in conditions of abnormal Fe utilization; |
||
|
long-term hemodialysis; injection of Fe prep- |
||
|
arations). |
|
|
|
Toxic cell damage (→A3) is the result of in- |
||
|
creased Fe deposition (especially in the form of |
||
|
hemosiderin [siderosis]). Participating in this |
||
|
are: 1) Fe-mediated formation of O2 radicals |
||
|
(lipid peroxidation of cellular membranes); 2) |
||
|
DNA damage; and 3) an increased formation of |
||
|
collagen, initiated by Fe. |
|
|
|
When the Fe content in the liver has in- |
||
|
creased to ca. 20 times the normal amount, fi- |
||
|
brosis |
with subsequent cirrhosis develops |
|
|
(→p.172ff.). The risk of lethal hepatocellular |
||
|
carcinoma increases twohundredfold. Pan- |
||
|
creatic fibrosis caused by siderosis results in |
||
|
insulin deficiency and thus diabetes mellitus. |
||
|
The incorporation of melanin and hemosiderin |
||
|
in the skin (especially if exposed to the sun) |
||
|
leads to marked pigmentation (“bronzed dia- |
||
|
betes”). Siderosis in the heart causes a cardio- |
||
|
myopathy that through arrhythmia and heart |
||
252 |
failure is a frequent cause of death in young |
||
patients. The joint damage (pseudogout) is in |
|||
|
part due to vitamin C deficiency brought about |
||
by Fe (Fe accelerates ascorbic acid breakdown).
Wilson’s Disease
Copper (Cu) metabolism (→B). Normal Cu uptake is ca. 2 – 5 mg daily, of which 40– 60% are absorbed in the stomach and upper duodenum. It is then incorporated mainly into ceruloplasmin (CP) in the liver. It reaches the systemic plasma in this form (ca. 93% of plasma Cu; →B1). CP, which binds six to seven Cu atoms relatively firmly, is apparently important for the oxidation of Fe2+ in plasma (→p. 38), but only a little CP-bound Cu is released into the tissues. This is not true of the portion of Cu that is bound to transcuprin and albumin (ca. 7% of plasma Cu). Old (desialysed) CP is broken down in the liver and the liberated Cu is excreted, firmly bound to biliary proteins (→B2), in the bile and stool (ca. 1.2 mg/d).
Wilson’s disease (hepatolenticular degeneration) is an autosomal recessive disorder of Cu metabolism in which the liver, central nervous system, eyes, and other organs are overloaded with Cu. The imbalance is caused by genetic defects fo the Cu-transporting Cu-ATPase. The defect results in failure to excrete sufficient Cu via the bile, the normal route, and the ability to incorporate Cu into CP is diminished (→B). As a result, free or only loosely bound Cu accumulates in the liver and then in plasma (at subnormal total Cu concentration) and in other organs (→B3). In this form it is cytotoxic because it binds to proteins, especially the sulfhydryl groups, and promotes the formation of O2 radicals (lipid peroxidation). The effects (→B4) are hemolytic anemia and chronic active hepatitis that can later change to cirrhosis. If the hepatitis takes a fulminant course, large amounts of Cu are suddenly released from the necrotic liver and this may trigger a hemolytic crisis. The accumulation of Cu in the CNS can cause numerous and diverse neurological, neuromuscular, and psychogenic abnormalities. Deposition of granular Cu in Descemet’s membrane of the eye can bring about a Kayser–Fleischer ring around the periphery of the cornea. The kidneys, skeleton, and heart can also be affected.
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.
A. Hemochromatoses
Primary |
1 |
|
|
|
|
|
|
|
hemochromatosis |
|
Secondary hemochromatoses |
2 |
|
|
|
||
inherited autosomal |
|
|
|
Disease |
||||
recessive disease |
|
ineffective erythropoiesis, chronic |
|
|
||||
(Cys-282-Tyr mutation |
liver diseases, porphyria cutanea tarda, |
|
|
|||||
in 83% of patients) |
|
parenteral Fe overloading, etc. |
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
Raised Fe absorption |
|
|
|
|
Wilson’s |
||
|
|
|
|
|
|
|
|
|
|
|
Hemochromatosis |
|
|
Venesection |
|
Hemochromatosis, |
|
|
Fe content |
(from 2 – 6 g to > 50 g!) |
|
|
(0.5 g Fe/L of blood) |
|||
|
|
|
|
Treatment |
|
|
||
Fe toxicity: lipid peroxidation, DNA damage, collagen formation |
3 |
|||||||
|
|
|
|
|
|
|
||
|
|
|
|
|
Heart: |
Joints: |
|
|
|
|
|
|
|
Vitamine C |
|
||
Liver: |
|
|
|
|
|
|
||
|
|
|
|
|
|
8.6 |
||
|
|
|
|
Siderosis |
deficiency |
|
||
|
Pancreas: |
|
|
|
|
|||
Siderosis |
|
Skin: |
|
|
|
|||
Siderosis |
|
Cardiomyopathy |
|
|
Plate |
|||
Fibrosis |
|
Incorporation of |
Pseudogout |
|
||||
|
|
|
|
|
||||
|
|
|
|
|
|
|||
|
Fibrosis |
|
melanin and |
|
|
|
|
|
Cirrhosis |
|
hemosiderin |
Arrhythmia |
|
|
|
||
|
|
|
|
|
||||
|
|
|
Testes: |
|
|
|||
Diabetes mellitus |
|
|
|
|
|
|||
Pigmentation |
|
Heart failure |
Infertility |
|
|
|||
Carcinoma |
|
|
|
|
|
|||
B. Wilson’s disease |
|
|
||
|
|
Gene defect (chromosome 13): |
Biliary |
3 |
|
|
Wilson’s disease |
|
|
|
|
Cu elimination |
||
|
|
|
||
Cu |
|
Ceruloplasmin |
Cu incorporation in |
|
|
|
|||
|
|
|
||
|
|
|
ceruloplasmin |
|
|
|
Cu intake: |
|
|
|
|
2 – 5 mg/d |
Free Cu |
|
|
|
Protein |
||
|
|
Cu |
|
|
|
|
absorption: |
|
|
|
|
40 – 60 % |
|
|
Liver |
|
|
Chronic |
|
|
|
Stomach |
|
|
|
|
hepatitis |
|
|
Plasma |
|
Duodenum |
|
Cornea |
|
|
|
||
|
|
|
|
|
|
|
|
4 |
|
1 |
|
2 |
Cirrhosis |
|
|
Transcuprin |
|
Neurological |
|
|
|
disorders |
||
|
|
Cu excretion |
Hemolysis |
|
|
|
|
||
|
|
ca. 1.2 mg/d |
253 |
|
|
Albumin |
|
||
|
Tissue |
|
||
|
|
|
|
|
Photo: Hollwich F. Taschenatlas der Augenheilkunde. 3rd ed. Stuttgart: Thieme: 1987.
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.

8 Metabolism
254
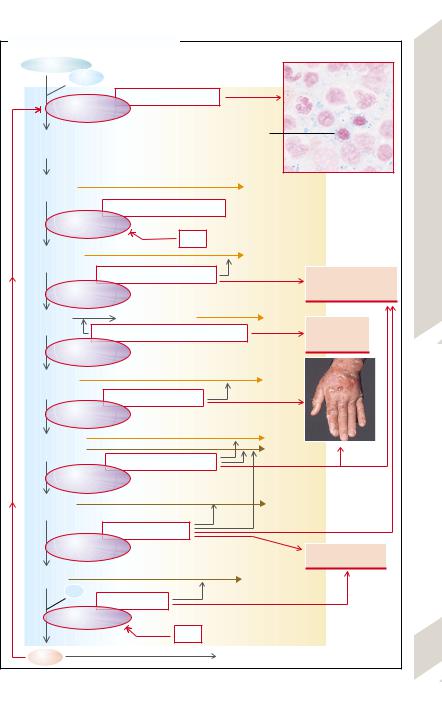
Heme Synthesis, Porphyrias
Heme is synthesized in a chain of eight reactions (→A). In addition to its incorporation into the hemoglobin of the erythroblasts (→p. 39), heme is synthesized in practically all organs and built into myoglobin, cytochrome P450, catalase, peroxidase, or the re- spiratory-chain cytochrome. Because these hemoproteins are indispensable, complete absence of heme synthesis is incompatible with life. Partial, usually heterozygous, defects of one of the participating enzymes have severe consequences.
Heme synthesis starts with the formation of α-amino-β-ketoadipate that is spontaneously transformed into δ-aminolevulinate (δ-amino- levulinic acid [δ-ALA]). This step, which takes place in the mitochondria, is the rate limiting step of heme synthesis; it is catalyzed in the erythroblasts by δ-ALA synthetase 2 (→A1) and in the liver by δ-ALA -synthetase 1. The activity of both isoenzymes is reduced by heme, the end-product of the synthesis (negative feedback; →A, left). This happens in part through heme being bound in the cytosol to the heme-regulated element of the proenzyme and hindering the latter from passing into the mitochondria.
Effects of heme synthesis abnormalities differ depending on this feedback, depending on whether the substrate turnover of δ-ALA-syn- thetase-2 or of one of the subsequent enzyme reactions is reduced. In the former case (→A1), the heme deficiency can only inadequately raise the activity of the deficient δ- ALA–synthetase-2, so that a sideroblastic anemia will develop (→p. 36).
In deficiencies of the follow-on enzymes (→A2 –8) a hugely increased availability of δ- ALA (disinhibition of δ-ALA-synthetase) develops due to the intact negative feedback. As a result, the concentrations of the substrates of all subsequent reactions are increased and thus the turnover is increased until enough heme has been produced. It is the high concentrations of the intermediary substances that lead to abnormalities (primary porphyrias; →A2 –8). Depending on their solubility in water or lipids, the intermediary products are excreted in the urine (δ-ALA, porphobilinogen [PBG]), uroporphyrin), or additionally via the
bile in the stool (coproporphyrins, protoporphyrins), respectively. The porphyrins are produced from the respective porphinogens; their excretory pattern is of diagnostic significance.
The concentration of δ-ALA is raised by a deficiency of δ-ALA-dehydratase (= PBG synthetase) (→A2) as well as by a hypofunction of porphobilinogen deaminase (also called hydroxymethylbilane synthetase), the cause of acute intermittent porphyrias (→A3), in which the PBG concentration is also increased. This results in neurovisceral dysfunctions
(tachycardia, nausea, vomiting, constipation) and neuropsychogenic disorders (paralyses, seizures, coma, hallucinations). One of the causes of these dysfunctions may be the competition between δ-ALA and the structurally similar neurotransmitter γ-aminobutyrate (GABA).
In congenital erythropoietic porphyria
(→A4) uroporphyrinogen will be formed nonenzymatically from hydroxymethylbilane, and converted enzymatically to coproporphyrinogen I (analogously to A5). Coproporphyrinogen I can no longer be used metabolically and, excreted in the urine, in infants it causes red stains on diapers and later on the teeth. Other effects are skin reactions to light and hemolytic anemia.
In (the more frequent) porphyria cutanea tarda (→A5) the porphyrins cause damage to the skin (poorly healing blisters; →A, photograph) as a result of light absorption (especially at λ= 440 nm). O2 radicals are involved in the generation of the skin lesions.
In hereditary coproporphyria (→A6), as also in porphyria variegata (→A7) (particularly common in South Africa [ca. three out of every 1000 whites]), δ-ALA, PBG, and the coproporphyrins are all elevated, causing neuropsychogenic and dermatological symptoms in the affected children. In protoporphyria (increase of protoporphyrin; →A8) burns, itching, and pain in the skin due to photosensitivity are prominent after exposure to ultraviolet rays.
Acquired porphyrias occur in lead poisoning
(→A2,8; high δ-ALA and PBG levels) and in hepatobiliary diseases, in which coproporphyrin excretion in bile is reduced.
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.
A. Abnormalities of Heme Synthesis |
|
|
|
||
Succinyl-CoA |
|
|
|
|
|
+ glycine |
|
|
|
|
|
δ-ALA- |
|
Sideroblastic anemia |
|
|
|
|
|
|
|
|
|
synthase-2 |
|
1 |
Circular |
|
|
|
|
|
|
Porphyrias |
|
α-amino- |
|
Fe incorporation (blue) |
|
||
|
in erythroblasts |
|
|||
β-ketoadipate |
|
|
|
|
|
δ-amino- |
|
|
|
|
|
|
|
in urine |
|
Synthesis, |
|
levulinate |
|
|
|
||
|
|
|
|
||
δ-ALA-dehydratase deficiency |
|
|
|||
δ-ALA- |
|
2 |
|
|
|
dehydratase |
|
|
|
||
|
|
Lead |
|
|
Heme |
Porphobili- |
|
|
in urine |
|
|
nogen (PBG) |
|
|
|
||
|
|
|
|
||
|
|
|
|
|
|
Acute intermittent porphyria |
|
Neurological and |
8.7 |
||
PBG |
|
3 |
|
psychogenic |
|
|
|
disorders |
Plate |
||
desaminase |
|
|
|||
|
|
|
|
||
Hydroxy- |
Uroporphyrinogen I |
in urine |
|
||
Hemolytic |
|
||||
methylbilane |
|
|
|
|
|
Congenital erythropoietic porphyria |
|
anemia, |
|
||
Uroporphyrinogen- |
4 |
|
red teeth |
|
|
|
|
|
|||
III-cosynthase |
|
|
|
|
|
|
|
|
|
|
|
Uroporphy- |
|
|
in urine |
|
|
rinogen III |
|
|
|
|
|
|
|
|
|
|
|
Porphyria cutanea tarda |
|
G. Krämer |
|
||
Uroporphyrinogen |
5 |
|
|
||
decarboxylase |
|
|
|
||
Copropor- |
|
|
in urine |
Photo: |
|
|
|
|
|
||
phyrinogen III |
|
|
in stool |
|
|
Hereditary coproporphyria |
|
|
|
||
Coproporphyrino- |
6 |
|
|
|
|
gen-III-oxidase |
|
|
|
|
|
Protopor- |
|
|
in stool |
|
|
phyrinogen |
|
|
|
|
|
|
|
|
|
|
|
Porphyria variegata |
|
|
|
||
Protoporphyrino- |
|
7 |
|
Marked |
|
gen oxidase |
|
|
|
||
|
|
|
|
photosensitivity |
|
Protopor- |
|
|
in stool |
|
|
phyrin |
|
|
|
|
|
|
|
|
|
|
|
+ Fe |
|
|
|
|
|
Protoporphyria |
|
|
|
||
Ferrochelatase |
|
8 |
|
|
|
|
|
Lead |
|
|
|
Heme |
|
Hemoglobin and other heme proteins |
255 |
||
|
|
||||
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.

9 |
Hormones |
F. Lang |
 General Pathophysiology of Hormones
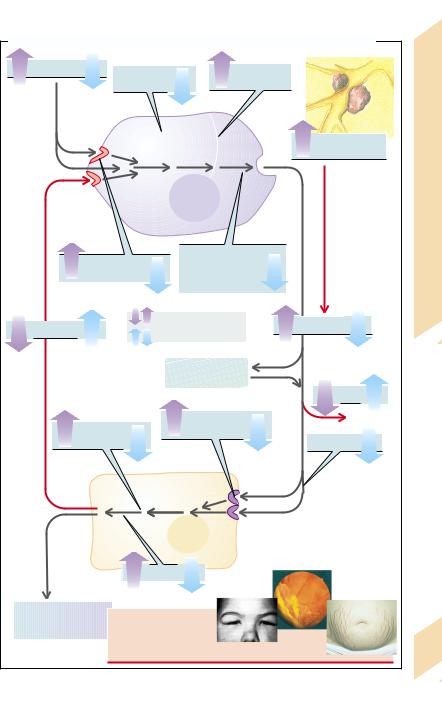
General Pathophysiology of Hormones
Hormones serve to regulate and to control organ functions. Their release is dependent on stimulation (or inhibition) through specific factors. Hormones act upon hormone-produc- ing cells themselves (autocrine), they influence neighboring cells (paracrine), or they reach target cells in other organs via the blood (endocrine). In a stricter sense, hormones achieve their effects predominantly via an endocrine path. For endocrine action to be effective the hormones must not be inactivated before reaching their target cells. Some hormones require activation (see below). The transition from endocrine hormones to paracrine mediators and transmitters is a fluid one.
At the target cells the hormones bind to receptors and exert their effects via various mechanisms of cellular signal transduction. It is usually through a reduction of stimulating factors that these effects lead to a reduced release of the particular hormone, i.e., there is a regulating cycle with negative feedback
(→A6). In a few cases there is positive feedback (of limited duration), i.e., the hormones cause an increased activity of stimuli and thus promote their own release. The term controling (→A1) is used when hormone release is influenced independently of hormonal effects. Several independent controling and regulating stimuli can act on the hormone-producing glands.
A reduced hormone effect (blue arrows) can be due to abnormal hormone synthesis and storage. Other causes can be abnormalities of transport within the synthesizing cells or abnormalities of release (→A5). Hormone deficiency may also arise when the hormonal glands are not stimulated sufficiently to meet the needs of the organism (→A1), when the hormone-producing cells do not react sensitively enough to the stimuli (→A4), or when there are not enough hormone-producing cells (hypoplasia, aplasia; →A2).
Other possible causes are too rapid inactivation or accelerated breakdown of hormones. In the case of hormones that are bound to plasma proteins (→A7) the duration of action de-
256pends on the proportion of bound hormones. In their bound form hormones cannot exert
their effect; on the other hand, they escape being broken down or being excreted by the kidney.
Some hormones must first be converted into their effective form at the site of their action (→A8). However, if this conversion is not possible, for example, due to enzyme defects, the hormone will have no effect. Hormonal action may also not occur because the target organ is unresponsive (e.g., due to defective hormone receptors or faulty intracellular transmission) or functional incapacity of the target cells or organs (→A9).
Causes of increased hormone effects (violet arrows) include, first of all, increased hormonal release. This may be due to an excessive influence of individual stimuli (→A1), increased sensitivity (→A4), or too large a number of hormone-producing cells (hyperplasia, adenoma; →A2). Hormonal excess can also be caused by the production of hormones in undifferentiated tumor cells outside of hormonal glands (ectopic hormonal production; →A3). The small-cell bronchial carcinoma is particularly frequently active endocrinally.
Raised hormonal action is also to be expected if a hormone is broken down or inactivated too slowly (→A7; e.g., in dysfunction of the inactivating organ [kidney or liver]). The breaking down can be delayed by binding to plasma proteins, but the protein-bound proportion would not be exerting any action either (see above).
Finally, hormonal effects can be increased by hypersensitivity of the target organ (too many hormone receptors or ones that are too sensitive), by increased intracellular transmission, or hyperfunction of the hormone-sensi- tive cells (→A9).
The clinical features, i.e., the sum of the pathophysiological changes in the organism, are the result of reduced or increased hor- mone-specific effects.
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.
A. Excess and Deficiency of Hormones as Disease-Producing System (Overview) |
|
|||||
Regulating influences |
|
|
2 |
|
|
|
Hypoplasia, |
|
Hyperplasia, |
of Hormones |
|||
|
|
|
||||
1 |
|
aplasia |
|
adenoma |
|
|
|
|
|
|
|
||
|
|
|
|
|
Ectopic |
|
|
|
|
|
|
Pathophysiology |
|
|
|
|
|
|
hormone production |
|
|
|
|
|
|
3 |
|
|
|
Hormon-producing |
|
|||
|
|
glandular cell |
5 |
|
General |
|
|
|
|
|
|
||
|
|
4 |
Hormone synthesis, |
|
||
|
Receptors: |
|
||||
|
|
intracellular |
|
|||
|
|
|
|
|||
|
density, affinity |
|
transport, |
|
9.1 |
|
|
|
|
|
release |
|
|
|
|
|
|
|
Plate |
|
|
|
|
|
|
|
|
|
|
Increased hormonal effect |
Hormone concentration |
|
||
Feedback |
|
Decreased hormonal effect |
|
|||
|
|
|
||||
6 |
|
|
|
|
7 |
|
|
|
|
|
Binding to |
|
|
|
|
|
plasma proteins |
|
|
|
|
|
|
|
|
Breakdown |
|
Intracellular |
|
Receptors: |
|
|
||
|
density, affinity |
|
|
|||
transmission |
|
|
Conversion |
|
||
|
|
|
|
|
8 |
|
9 |
|
|
|
|
|
|
|
|
|
|
Target |
|
|
|
|
|
|
cell |
|
|
|
|
Cell function |
|
|
||
Hormonal effect |
|
|
|
|
|
|
|
|
Clinical features of endocrinal disease |
257 |
|||
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.

9 Hormones
258
Abnormalities of Endocrinal Regulatory Circuit
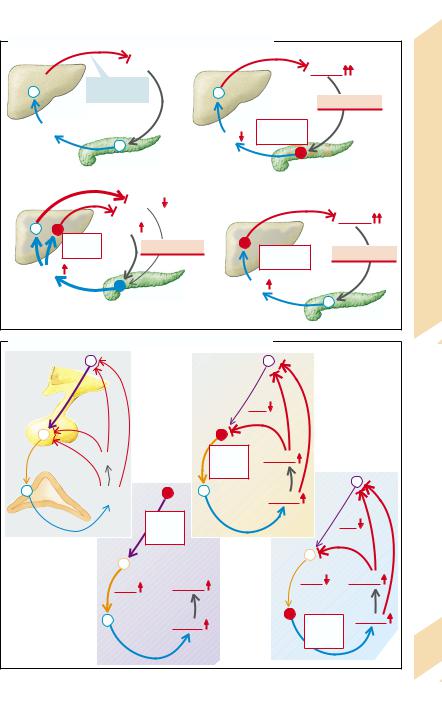
Hormones are usually part of regulatory circuits. Disorder of one element in such a circuit leads to characteristic changes in its other elements.
Pituitary-independent release of hormones is usually regulated by those parameters that are influenced by the particular hormone, the latter acting on the target organs, whose functions in turn lead to a reduction in the stimuli leading to hormone release (regulatory circuit with negative feedback). Insulin release serves as an example (→A1): raised plasma glucose concentration stimulates the release of insulin, the effect of which on the target organ, for example, the liver (increased glycolysis; inhibition of gluconeogenesis and glycogen formation), leads to a reduction in plasma glucose concentration.
If the insulin release is inappropriately high for a given plasma glucose concentration (hyperinsulinism), this will lead to hypoglycemia. In addition to an insulin-producing tumor, the cause may be an overlap of regulatory circuits, in that some amino acids also stimulate insulin release, and some of the insulin effects (stimulation of protein synthesis, inhibition of proteolysis) can produce a reduction in the plasma concentration of amino acids. An abnormal breakdown of amino acids, for example, one due to an enzyme defect, can trigger hypoglycemia via a rise in amino acid concentration in the blood and subsequent stimulation of insulin release (→A2).
If there is a defective hormonal gland
(→A3), the hormone level and thus the hormonal effect is reduced. In the example illustrated an insufficiency of the beta cells results in hyperglycemia.
In addition, when the responsiveness of the target organs is reduced (→A4), the hormonal effect is decreased. In this way liver failure can result in hyperglycemia, which in turn will raise plasma insulin concentration. However, the abnormal breakdown of amino acids in liver failure can cause hypoglycemia through hyperaminoacidemia and by stimulating insulin release (see above; →A2).
Hormone release regulated by the hypothalamus and pituitary. The plasma concentration of hormones that are under the influence of the hypothalamus and pituitary gland is always regulated (→B1). Liberins (releasing hormones), formed in the hypothalamus, cause the release of tropins in the pituitary. These stimulate the release of the respective hormone in the periphery. The hormone and to some extent also the effect produced by the hormone finally inhibit the release of liberins in the hypothalamus and of tropins in the pituitary. The example illustrates the regulation of cortisol from the adrenal cortex.
Reduced release of peripheral hormones may be due to a loss of function in the hypothalamus, pituitary, or peripheral hormonal gland. The primary cause of an increased release of peripheral hormones can be an inadequately high orthotopic or ectopic release (→p. 257 A3) of liberins, tropins, or peripheral hormones.
If there is an increase in liberin release
(→B2), liberin, tropin, and peripheral hormone concentrations are raised.
If there is a primary increase in tropin release, the concentrations of tropins and of the peripheral hormone will be raised, but that of liberins reduced (→B3).
If there is a primary rise in peripheral hormone release, the release of liberins and tropins is suppressed (→B4).
In an analogous manner, a primary deficiency of liberins will lead to tropin and peripheral hormone deficiency, while a primary lack of tropins will result in a reduced release of peripheral hormones, with increased release of liberins; a primary deficiency of peripheral hormones will lead to increased release of liberins and tropins.
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.
A. Abnormalities of Simple Endocrinal Regulatory Circuit |
|
|
|
|
|||
|
Glucose |
|
Glucose |
|
|||
Glycolysis |
|
|
|
|
|||
Negative |
|
|
|
|
|
|
|
etc. |
|
|
|
|
|
|
|
|
feedback |
|
|
Hyperglycemia |
|
||
Liver |
|
|
|
||||
|
|
|
|
|
|
||
Insulin |
|
Insulin |
Beta-cell |
|
|
|
Circuit |
|
|
insufficiency |
|
|
|
||
|
|
|
|
|
|
|
|
1 |
Pancreas |
|
|
|
|
Regulatory |
|
|
|
|
|
|
|
||
Normal regulation |
3 |
|
|
|
|
||
|
|
Defect in hormonal gland |
|
|
|||
|
Glucose |
|
|
|
|
||
|
Amino |
|
|
|
|
Glucose |
Endocrinal |
|
acids |
|
|
|
|
|
|
|
Enzyme |
Hypoglycemia |
|
|
|
|
|
|
defect |
Liver |
|
Hyperglycemia |
|||
|
|
|
|||||
|
|
|
insufficiency |
|
|
||
Insulin |
|
|
|
|
|
||
|
|
|
|
|
|
9.2 |
|
|
|
Insulin |
|
|
|
||
2 |
|
|
|
|
|
|
Plate |
Increased secretion in the |
4 |
|
|
|
|
||
intertwined regulatory circuit |
Target organ defective |
|
|
|
|
||
B. Abnormalities of Hypothalamus-Regulated Hormones |
|
|
|
|
|||
Hypothalamus |
|
|
|
|
|
|
|
1 |
|
|
|
|
|
|
|
|
Liberin |
|
CRH |
|
|
|
|
Pituitary |
(CRH) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
gland |
|
|
|
|
|
|
|
Tropin |
Effect |
ACTH |
Effect |
|
|
|
|
primarily |
|
|
|
|
|||
(ACTH) |
|
|
|
|
|
|
|
|
|
raised |
|
|
|
|
|
|
Hormone |
3 |
|
|
|
|
|
|
|
Cortisol |
|
|
|
|
|
|
(cortisol) |
|
|
|
|
|
|
Adrenal |
|
CRH |
|
|
|
CRH |
|
cortex |
|
primarily |
|
|
|
|
|
|
|
|
|
|
|
||
Normal regulation |
raised |
|
|
|
|
|
|
2 |
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
ACTH |
Effect |
ACTH |
|
Effect |
|
|
|
|
|
|
|
|
||
|
|
|
|
4 |
|
|
|
|
|
Cortisol |
|
Cortisol |
Cortisol |
|
|
|
|
|
|
|
|||
|
|
|
|
primarily |
|
||
|
|
|
|
raised |
|
|
259 |
|
|
|
|
|
|
|
|
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.

9 Hormones
260
The Antidiuretic Hormone
The antidiuretic hormone ([ADH] adiuretin, vasopressin) is formed in the supraoptic and paraventricular nuclei of the hypothalamus and is transported to the posterior lobe of the pituitary gland via the axons of the hormoneproducing neurons. ADH causes the incorporation, via V2-receptors and cAMP, of water channels into the luminal membrane and thus promotes water reabsorption in the distal tubules and in the collecting duct of the kidney. ADH also stimulates the tubular absorption of Na+ and urea. A high ADH concentration also leads to vasoconstriction (via V1-receptors and IP3).
Stimuli for the release of ADH are extracellular hyperosmolarity (or cell shrinkage) and a decreased filling of the two atria, as well as vomiting, pain, stress, and (sexual) arousal. ADH secretion is further stimulated by angiotensin II, dopamine, and some drugs or toxins (e.g., nicotine, morphine, barbiturates). Increased atrial distension as well as γ-aminobu- tyric acid (GABA), alcohol, and exposure to cold have an inhibitory effect.
ADH excess (→ A1) is often due to raised ADH formation in the hypothalamus, for example, by stress. Furthermore, ADH can be formed ectopically in tumors (especially small-cell bronchial carcinoma), or by lung disease. It leads to reduced water excretion (oliguria). The resulting marked concentration of poorly dissolved urinary constituents can lead to the formation of urinary stones (urolithiasis). At the same time there will be a drop in extracellular osmolarity (hypotonic hyperhydration) and cell swelling occurs. This is especially dangerous if it leads to cerebral edema (→ p. 358).
ADH deficiency (→ A2) occurs if release is reduced, as in genetically determined central diabetes insipidus, in destruction of neurones, for example, by autoimmune disease, or other pituitary gland injury. Exogenous causes include alcohol or exposure to cold. On the other hand, ADH may fail to have an effect on the kidney, even if it is normally secreted, for example, because of defective water channels, or if the concentrating capacity of the kidney is otherwise impaired, as in K+ deficiency, Ca2+ excess, or inflammation of the renal medulla (renal diabetes insipidus). Decreased ADH re-
lease or effect results in the excretion of large amounts of poorly concentrated urine and hypertonic dehydration (see also p.122), leading to cell shrinkage. Patients will be forced to compensate for the renal loss of water by drinking large amounts (polydipsia). If the osmoreceptors in the hypothalamus are destroyed, ADH deficiency is accompanied by hypodipsia, and the hypertonic dehydration is especially marked. In psychogenic polydipsia
ADH release is inhibited because of the excess water, and thus, contrary to primary ADH deficiency, the result will be hypotonic hyperhydration.
Prolactin
Prolactin (→ B) is formed in the anterior lobe of the pituitary gland. It stimulates growth and differentiation of the mammary gland as well as milk production. It inhibits the pulsatile, but not the basal, release of the gonadotropins (luteinizing hormone [LH] and follicle-stimu- lating hormone [FSH]; → p. 274). It also inhibits cellular glucose uptake and the cellular immune defenses.
Touching the nipple of a lactating woman and estrogens promote the release of prolactin. Its release is also stimulated by thyroliberin (thyroid-releasing hormone [TRH]), endorphins, vasoactive intestinal peptide (VIP), oxytocin, and angiotensin II as well as by stress, non–rapid eye movement (NREM) sleep, or hypoglycemia. Dopamine inhibits prolactin release. As prolactin increases dopamine metabolism in the hypothalamus, it inhibits its own release (negative feedback).
Excess prolactin (→ B) can be caused by hor- mone-producing tumors, or by administration of antidopaminergic drugs. Renal and liver failure can also result in an excess of prolactin, possibly via a lack of dopamine. Hypothyroidism raises prolactin release via correspondingly increased TRH secretion. The effects of excess prolactin are milk flow (galactorrhea), tendency toward hyperglycemia, and an inhibition of gonadotropin release, accompanied by hypogonadism, amenorrhea, loss of libido, and impotence.
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.