

книги студ / Color Atlas of Pathophysiology (S Silbernagl et al, Thieme 2000)
.pdfA. General Functional Disorders
Receptor |
ACh |
Ion channels blocked |
|
||
density |
|
||||
Receptor |
|
|
|
Extracellular: K+ |
Noxious factor |
blockade |
|
|
+ |
||
|
G |
|
K |
|
|
|
|
|
|
|
|
|
|
+ |
|
Na+ Na+/K+-ATPase |
|
G-protein |
|
K |
|
inhibited |
|
|
|
|
|
|
|
blocked |
|
|
|
|
|
1 |
Functional disorders |
|
|
|
|
|
|
Axonal transport |
|
|
|
Necrosis |
Apoptosis |
||
|
|
|
|
|
|
|
|
|
Neuro- |
|
|
|
|
|
|
Formation |
transmitter |
|
|
|
Cell death |
3 |
|
Storage |
|
|
|
||||
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
Release |
N |
Reversible |
Irreversible |
|
|||
|
|
|
|
Partial |
|||
|
T |
|
|
|
|
||
|
|
|
|
|
compensation |
||
|
|
|
|
|
|
||
2 |
Inactivation |
|
|
|
|
|
|
|
|
Functional deficits |
|
||||
|
|
|
|
||||
B. Blood-Brain Barrier |
C. Axon Transection and Regeneration |
|
|||||
|
Retrograde |
|
|
|
|
|
|
Blood-brain |
transneuronal |
|
|
|
|
|
|
degeneration |
|
|
|
|
|
||
barrier |
|
|
|
|
|
|
|
|
Chromatolysis |
|
|
|
|
|
|
Toxins |
|
|
|
|
|
|
|
Stimuli |
|
|
|
|
Thirdly: |
|
|
Immune-competent |
Lesion |
|
|
|
Schwann cell |
|
|
|
|
|
|
takes over |
|
||
cells, antibodies |
|
Nerve |
|
|
|||
|
|
NGF synthesis |
|||||
|
|
Growth |
|
||||
|
|
|
|
|
|
||
Retrograde axonal |
|
Factor |
|
Interleukin1 |
|
||
transport: 1m/day |
Myelin |
|
|
|
|
||
|
|
|
|
|
Secondly: |
||
|
disintegration |
|
|
|
macrophage |
||
|
Waller |
|
|
|
|
stimulates |
|
|
|
|
|
|
Schwann |
||
|
degeneration |
|
|
|
|||
|
|
|
|
|
cell |
||
|
Anterograde |
|
|
|
|
||
|
|
|
|
|
|
||
|
transneuronal |
|
|
|
Firstly: |
|
|
Pertussis |
degeneration |
|
|
|
retrograde |
|
|
toxin etc. |
|
|
|
|
|
NGF transport |
|
Viruses |
Target cell |
|
|
|
|
interrupted |
|
1 |
|
|
|
|
|
|
|
Organic heavy |
|
|
2 |
|
|
|
|
Effects of axon |
|
|
|
|
|
||
metal compounds |
|
|
Regeneration in |
|
|||
transection |
|
|
|
||||
|
|
|
peripheral nerve |
|
|||
|
|
|
|
|
|||
Plate 10.2 Pathophysiology of Nerve Cells
301
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.

10 Neuromuscular and Sensory Systems
302
Demyelination
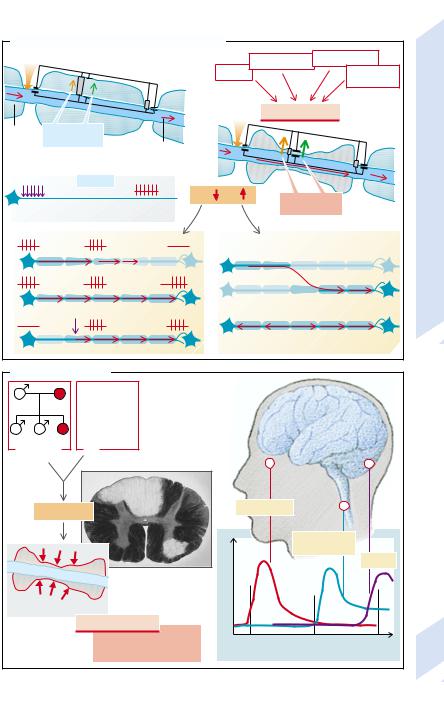
In myelinated nerves, the axon between two nodes of Ranvier (internodal segment) is surrounded by a myelin sheath (→A). This is a precondition for saltatory conduction of the action potentials, i.e., the “jumping” propagation of excitation from one nodal constriction (R1) to the next (R2). The internodal segment itself cannot generate an action potential, i.e., depolarization of the second node (R2) is completely dependent on the current from the first node (R1). However, the current is usually so strong that it can even jump across the nodes.
Nevertheless, on the way along the internodal segment the amplitude of the current will diminish. First of all, the membrane in the internodal segment must change its polarity, i.e., the membrane capacitance must be discharged, for which a current is needed (→A, green arrow). Secondly, current can also escape through individual ionic channels in the axonal membrane (orange arrow). However, myelination of the internodal segment causes the membrane resistance (Rm) to be elevated and the capacity (Cm) of the membrane condensor to be reduced (→A, left).
The resistance of the axonal membrane of the internodal segment is very high because of the low density of ionic channels there. Furthermore, the perimembranous space is insulated by a layer of fat from the free extracellular space. The low capacitance of the condensor is due to the large distance between the interior of the axon and the free extracellular space as well as the low polarity of the fatty material in the space between them.
Demyelination (→A, right) can be caused by degenerative, toxic, or inflammatory damage to the nerves, or by a deficiency of vitamins B6 or B12. If this happens, Rm will be reduced and Cm raised in the internodal segment. As a result, more current will be required to change the polarity of the internodal segment (green arrow) and, through opening up the ionic channels, large losses of current may occur (orange arrow).
If, after the losses in the internodal segment, the current generated at R1 is not adequate to depolarize R2 to the threshold level, excitation is interrupted, even though the axon is completely intact. High frequency of
action potentials and low temperature favor interruption of conduction because of decreasing sensitivity of the node R2 (→A1). Minor lesions of the internodal segment can lead to slowing of conduction, because it can no longer jump across nodes and the next node has to be depolarized to its threshold before the excitation is passed to the afternext nodes (→A2). The resulting slowing may not be the same in different fibers, so that temporal dispersion of the signal may occur. Lastly, the damaged site may itself trigger action potentials, especially when the axon has concomitantly suffered spontaneous damage or is under mechanical pressure (→A3); excitation can jump across two neighboring damaged nerve fibers (ephaptic transmission; →A4), or conduction may run retrogradely (→A5).
Genetic defects of the myelin-sheath protein (e.g., protein O [P0, of peripheral myelin protein 22 (PMP 22)]) or of gap junctions in the Schwann cells (connexin 22) lead to certain hereditary peripheral neuropathies (Char- cot–Marie–Tooth syndrome, Dejerine–Sottas syndrome, Pelizaeus–Merzbacher disease).
The most important demyelinating disease is multiple sclerosis (→B). It is more common in women than men, familial aggregation sometimes occurs, and it has a higher incidence among carriers of HLA3 and HLA7. It is an autoimmune disease that may be triggered by a viral infection and is characterized by demyelinating inflammatory foci (→B1). The typical feature of multiple sclerosis is the temporally unrelated occurrence of completely different neuronal deficits, caused by lesions in different parts of the brain. Some of the lesions may partly regress when the local inflammatory process has subsided and the nerves (in the case of intact axons) have been remyelinated. The example in B2 illustrates that at first there is a fully reversible loss of vision due to a damaged optic nerve (→p. 326), followed by a partly reversible sensory loss when the sensory tracts of the spinal cord are affected (→p. 318). Finally, ataxia sets in when the cerebellum becomes involved (→p. 316).
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.
A. Development and Effects of Demyelination |
|
|
|
|
||||
Na+ |
Rm |
Cm |
Rm |
|
Inflammations Vitamin deficiency |
|
||
|
high |
low |
Toxins |
|
Genetic |
|
||
|
|
|
low Cm |
|
|
|||
|
|
|
high |
|
|
|
defects |
|
|
|
|
|
|
|
|
|
|
R1 |
|
|
|
|
|
Demyelination |
|
|
Myelin |
Small leakage |
R2 |
|
|
|
|
|
|
sheat |
currents |
|
Propaga |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
R1, R2 = Ranvier nodes |
|
action potential |
|
|
|
|
|
|
|
normal |
|
|
|
|
Demyelination |
||
|
|
|
Rm |
Cm |
|
Large leakage |
|
|
Stimuli |
|
Action potentials |
|
|
currents |
|
||
1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.3 |
|
|
|
|
4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
Plate |
|
|
|
|
|
|
|
|
|
|
|
|
5 |
|
|
|
|
|
3 |
|
|
|
|
|
|
|
|
B. Multiple Sclerosis |
|
|
|
|
|
|
||
|
+ |
Autoimmune |
|
|
|
|
|
|
|
disease, |
|
|
|
|
|
||
|
|
possibly |
|
|
|
|
|
|
|
|
caused by |
|
|
|
|
|
|
|
+ |
viruses |
|
|
|
|
|
|
|
? |
|
|
|
|
|
|
|
Genetic |
|
|
|
|
|
|
||
disposition |
|
|
|
|
|
|
|
|
|
Inflammation |
|
|
Loss of visus |
|
|
||
|
|
|
|
|
|
|
||
|
|
|
|
|
|
Sensory losses |
|
|
|
|
|
|
|
|
|
Ataxia |
|
|
|
|
Demyelinating foci |
Symptoms |
|
|
|
|
|
|
|
in spinal cord (after Netter) |
|
|
|
|
|
Demyelination in CNS |
|
|
|
|
|
|||
|
Multiple sclerosis |
|
|
|
|
|
||
1 |
|
|
periodically recurrent |
2 |
1992 |
1993 |
1994 |
|
|
|
|
neuronal losses |
Example of the course of the disease |
303 |
|||
|
|
|
|
|
||||
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.

10 Neuromuscular and Sensory Systems
304
Disorders of Neuromuscular Transmission
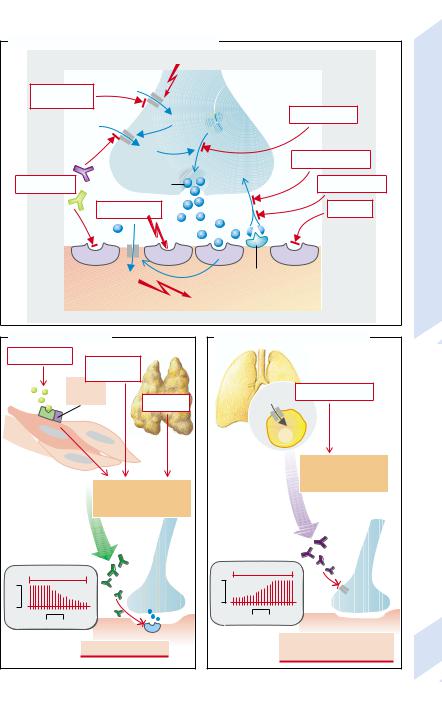
Neuromuscular transmission is a sequence of events (→A) that can be interrupted at various levels. The action potential is carried along by activation of the Na+ channels to the nerve ending, where it depolarizes the cell membrane and thus opens the voltage-gated Ca2+ channels. The Ca2+ ions that enter the nerve ending mediate the fusion of acetylcholine (ACh)-containing vesicles with the presynaptic membrane, whereupon ACh is released into the synaptic cleft. ACh binds to receptors of the subsynaptic membrane and in this way opens nonspecific cation channels. The depolarization of the subsynaptic membrane is transmitted to the postsynaptic membrane where, through opening of voltage-gated Na+ channels, an action potential is initiated that rapidly spreads over the entire muscle membrane. ACh is broken down by acetylcholinesterase; the choline which has been split off is again taken up into the nerve ending and used again for the synthesis of ACh.
Abnormalities can affect any element of this process. Local anesthetics, for example, inhibit the voltage-gated Na+ channels of the neuron and thus interrupt nerve transmission to the end-plate. The Ca2+ channels can be blocked by antibodies (see below). Botulinus toxin inactivates synaptobrevin, the protein responsible for binding the ACh-containing vesicles to the plasma membrane and thus for the release of ACh. The ACh receptors can also, like the Ca2+ channels, be blocked by antibodies which furthermore accelerate the internalization and breakdown of the receptors. The receptors can also be blocked by curare that, without itself having an effect, competitively inhibits the binding of ACh to the receptors.
Succinylcholine (suxamethonium chloride) leads to continuous stimulation of the receptors, continuous depolarization of the postsynaptic membrane, and thus to an inactivation of the postsynaptic Na+ channels. In this way it can, like curare, block neuromuscular transmission. In low concentrations, substances that inhibit acetylcholinesterase (e.g., physostigmine) increase neuromuscular transmission by increasing the availability of ACh in the synaptic cleft. In high doses, however, they inhibit neuromuscular transmission be-
cause high concentrations of ACh, as of succinylcholine, cause continuous depolarization of the subsynaptic membrane and so inactivate the postsynaptic Na+ channels. The re-up- take of choline into the nerve ending can be inhibited by Mg 2+ and hemicholine.
The most important disease affecting the end-plates is myasthenia gravis, a muscle paralysis that results from blockage of neuromuscular transmission (→B). It is caused by antibodies against the ACh receptors in the subsynaptic membrane which accelerate the breakdown of the receptors (→B1). This autoimmune disease can be caused by infection with viruses that have an ACh-receptor-like structure. Myasthenia may also occur in patients with a benign tumor of the thymus. The formation of such antibodies is favored in those who express special subtypes (DR3 and DQw2) of the major histocompatibility complex (MHC class II). In patients with myasthenia gravis, repetitive stimulation of a motor nerve will at first cause the production of a normal summated muscle action potential whose amplitude will, however, decrease through progressively increasing “fatigue” of neuromuscular transmission (→B2).
Another autoimmune disease that impairs neuromuscular transmission is the pseudomyasthenic syndrome of Lambert and Eaton
(→C). This condition often arises in patients affected by a small-cell carcinoma of the lung. Ca2+ channels in the plasma membrane of the tumor cells sensitize the immune system and stimulate the formation of antibodies that also react with the Ca2+ channels of the endplate (→C1). Due to inhibition of the Ca2+ channels, the summated muscle action potential is at first small, but is progressively normalized, because with the repetitive stimulation increasing amounts of Ca2+ are accumulated in the nerve endings (→C2).
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.
A. Disorders of Neuromuscular Transmission |
|
|
||
|
Action potential |
|
Disorders |
|
Local |
|
|
||
anesthetics |
|
|
||
|
|
Botulinus toxin |
||
|
Na+ |
Transmission: |
||
|
Ca2+ |
|
||
|
|
Mg2+, hemicholine |
||
Antibodies |
ACh |
Physostigmine |
||
Neuromuscular |
||||
Suxamethonium |
Curare |
|||
|
|
|
||
|
Na+ |
ACh-esterase |
10.4 |
|
Muscle cell |
|
|
||
Contraction |
Plate |
|||
|
||||
|
|
|
||
B. Myasthenia Gravis |
|
C. Pseudomyasthenic Syndrome |
|
|
Viral infection |
Genetic |
|
|
|
|
||
|
|
|
|
|
|
||
|
|
disposition |
|
|
|
|
|
|
DR3, |
|
|
|
|
Lung carcinoma |
|
|
DQw2 |
|
|
Ca2+ |
|
||
|
Thymoma |
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Sensitization |
|
|
|
|
|
|
|
against Ca2+channels |
|
|
|
|
Sensitization |
|
|
of the tumor cells |
|
|
|
|
|
Antibody |
|
|
|
|
|
|
against ACh receptor- |
|
|
|
|
|
|
|
like structure |
|
formation |
|
|
|
Antibody |
|
|
|
|
|
|
|
formation |
|
|
|
|
|
|
|
|
|
|
|
|
1 |
|
|
Repetitive stimulation |
|
1 |
|
Repetitive stimulation |
|
|
|
|
|
|
|
|
||
mV |
|
|
|
1mV |
|
|
|
1 |
|
|
|
|
|
|
|
2 |
50 ms |
|
|
2 |
50 ms |
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
Myasthenia gravis |
|
|
Lambert-Eaton |
305 |
||
|
|
|
myasthenic syndrome |
||||
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.

10 Neuromuscular and Sensory Systems
306
Diseases of the Motor Unit and Muscles
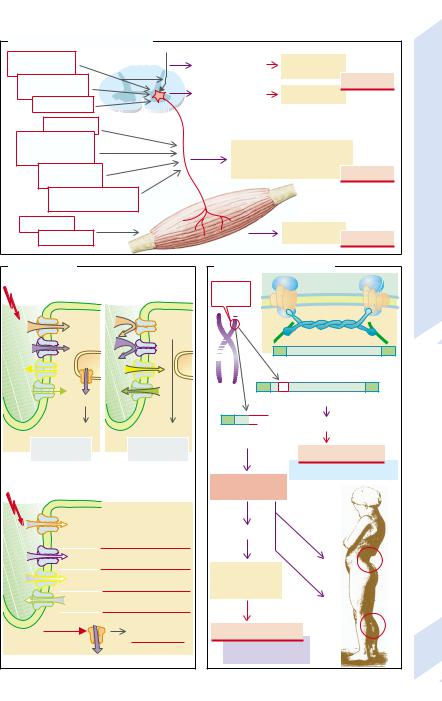
The motor unit consists of the motoneuron (α- motoneuron in the spinal cord or the cranial nerves), the associated axon, and all the muscle fibers innervated by its collaterals. The function of the motor unit can be affected by disease of the motoneuron, by interruption or delay of axonal conduction, or by disease of the muscle (→A).
The α-motoneurons can be infected by poliovirus and irreversibly destroyed by it. Also in spinal muscular atrophy, a group of degenerative diseases largely of unknown cause, these cells are destroyed. Amyotrophic lateral sclerosis (ALS) may be caused primarily by a disorder, partly genetic, of axonal transport that secondarily leads to the death of spinal α-motoneurons and supraspinal motoneurons (→A1).
Damage to or death of axons may, among others causes, be due to autoimmune diseases, a deficiency of vitamin B1 or B12, diabetes mellitus, poisoning (e.g., lead, alcohol), or genetic defects (e.g., Charcot–Maire–Tooth syndrome; →p. 302) (→A2).
The musculature (→A3) can also be affected by autoimmune diseases (e.g., dermatomyositis). In addition, genetic defects may involve the musculature, for example, in myotonia or dystrophy (see below).
Lesion of a motor unit causes paralysis of the affected muscles, regardless of whether it is localized in an α-motoneuron, axon, or the muscle itself (→A). In primary death of an α- motoneuron fasciculations typically occur. They are the result of synchronous stimulation and contraction of the muscle fibers of a motor unit. In ALS the destruction of the supraspinal neurons may result in hyperreflexia and spasticity (→p. 310), as long as some of the α-mo- toneurons are still intact. A lesion of a peripheral nerve which has reduced the thickness of the myelin layer will result in a slowing of the nerve’s conduction velocity (→p. 302). As a rule, sensory parts of the nerve are also affected. This leads to abnormal sensory functions as well as spontaneous action potentials in the damaged nerves, resulting in corresponding sensations (paresthesias). In primary death of muscles fibrillations often occur, i.e., uncoordinated contractions of individual muscle fibers.
Genetic ionic channel defects (→B) are the cause of a group of functional muscle diseases. Normally (→B1) depolarization of the muscle cell membrane is triggered on excitation by a voltage-gated Na+ channel that causes the opening of a voltage-gated Ca2+ channel (→ p. 304) and a Ca2+ channel of the sarcoplasmic reticulum. As a result, intracellular Ca2+ is increased, mediating muscular contraction. Repolarization is achieved by inactivation of the Na+ channels, by Cl– influx, and K+ efflux. This causes the inactivation of the Ca2+ channels so that the intracellular Ca2+ concentration again falls and the muscle relaxes.
Delayed inactivation of the Na+ channel due to mutation in the gene for the channel protein can lead to delayed relaxation, increased excitability, and cramps (Na+ channel myotonia and congenital paramyotonia; →B2). Cold further slows Na+ channel inactivation such that cramps occur, particularly in paramyotonia when the muscle gets cold. An additional defect of the Na+ channel or a defective K+ channel (?) can cause paralysis when the extracellular concentration of K+ is high (hyperkalemic periodic paralysis). A genetic defect of the volt- age-gated Ca2+ channel also leads to hypokalemic periodic paralysis. If there are defects in the Cl- channels, myotonia occurs. Depending on the severity of the molecular defect, inheritance of the disease is dominant (congenital myotonia, Thomsen’s disease) or recessive (Becker’s myotonia). In certain defects of sarcoplasmic Ca2+ channels the volatile halogenated anesthetics may bring about potential-inde- pendent activation of these channels with an increase in intracellular Ca2+. The resulting massively increased energy metabolism causes hyperthermia (malignant hyperthermia;
→p. 22).
In Duchenne’s or Becker’s degenerative muscular dystrophy (→C; →p. 307) dystrophin, an element of the cytoskeleton, is defective. The responsible gene is on the short arm of the X chromosome. The disease occurs practically only in males, because in females with one defective gene the dystrophin formed from the normal gene is sufficient. In Duchenne’s dystrophy only short, completely functionless dystrophin fragments are formed
!
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.
A. Diseases of the Motor Unit |
|
|
|
|
|||
Amyotrophic |
|
Death of supra- |
Hyperreflexia, |
|
|
||
lateral sclerosis |
|
spinal neurons |
spasticity |
|
|
||
|
Spinal |
|
|
Death of |
|
Paralysis |
|
1 |
muscular atrophy |
|
Fasciculation |
|
|
||
Poliomyelitis |
|
α-motoneurons |
|
|
|||
|
|
|
|
|
|||
|
|
|
|
|
|
||
|
Autoimmune |
α-motoneuron |
|
|
|
|
|
|
Metabolic |
|
|
|
|
I |
|
|
|
|
|
|
Muscles |
||
|
(B1, B12 |
deficiency, |
|
Slowing of conduction velocity, |
|||
|
diabetes mellitus) |
|
disorders of sensory functions |
|
|||
2 |
Toxic |
|
(sensory loss, paresthesias) |
Paralysis |
|||
|
(lead, alcohol) |
|
Axon |
|
|
and |
|
|
|
Genetic (Charcot- |
|
|
|
||
|
|
|
Muscle |
|
|
||
|
|
Marie-Tooth syndrome) |
|
|
Unit |
||
|
Autoimmune |
|
|
|
|
||
|
|
|
Fibrillations |
|
Motor |
||
|
3 |
Genetic |
|
|
Paralysis |
||
|
|
|
|
||||
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
B. Myotonias |
|
C. Muscular Dystrophies |
|
10.5 |
|||
Action potential |
1 |
Xp21 |
Sarcolemma |
|
|||
Repolarization |
|
||||||
|
Plate |
||||||
|
|
|
defect |
|
|
||
|
|
|
|
|
|
||
|
|
|
|
|
Muscle cell |
|
|
|
|
|
|
|
|
|
|
Na+ |
Intra- |
|
|
Dystrophin |
Actin |
|
|
|
|
cellular |
|
|
|
|
|
Ca2+ |
|
|
K+ |
|
|
|
|
|
Postsynaptic infolding |
K+ |
|
|
|
Normal dystrophin molecule |
|
|
|
|
Cl– |
|
|
Cl– |
2 |
|
|
|
|
|
Ca2+ |
|
|
1 |
Defective dystrophin |
|
|
|
Contraction |
|
Relaxation |
Nonfunctioning |
Disordered gait |
|
|
|
|
|
|
|
|
||||
Muscle |
|
|
Repolarization |
molecule |
|
|
|
|
cell |
Depolarization |
|
|
|
|
|||
|
|
|
|
|
||||
|
normal |
|
|
normal |
|
Becker’s dystrophy |
|
|
|
|
|
|
|
|
milder course of disease |
|
|
Action potential |
|
|
2 |
Severe |
|
|
|
|
|
|
muscle dystrophy |
|
|
|
|||
|
Na+ |
Na+ channel myotonia, |
|
|
|
|
||
|
congenital paramyotonia, |
Respiratory |
|
|
|
|||
|
|
|
periodic hyperkalemic |
|
|
|
||
|
|
|
muscle |
|
|
|
||
|
|
|
paralysis |
|
|
|
||
|
|
|
|
|
|
|
||
|
Ca2+ |
Periodic hypokalemic |
|
|
|
|
||
|
|
|
paralysis |
Respiratory |
Lordosis |
|
|
|
|
Cl |
– |
Congenital myotonia |
|
|
|||
|
failure, |
|
|
|
||||
|
|
(Thomsen, Becker) |
|
by Duchenne) |
|
|||
|
|
|
heart failure |
|
|
|||
|
+ |
|
Periodic hyperkalemic |
|
|
|||
|
|
|
|
|
||||
|
K |
|
paralysis |
|
Calf muscle |
|
||
|
Halothane Ca2+ |
Malignant |
|
hypertrophy |
|
|||
|
|
|
|
hyperthermia |
Duchenne’s dystrophy |
(Drawing |
|
|
|
|
|
|
|
20 years |
|
307 |
|
|
|
|
|
|
fatal course within |
|
|
|
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.

10 Neuromuscular and Sensory Systems
308
!
(→C1). The disease ends in death during the first 20 years of life. Hypertrophied yet weak calf muscles and marked spinal lordosis due to muscular weakness are typical for this form of dystrophy. In Becker’s dystrophy the dystrophin is also defective, but its function is less impaired and the disease therefore less severe (→C2; →p. 307).
Diagnosis of Motor Unit Diseases
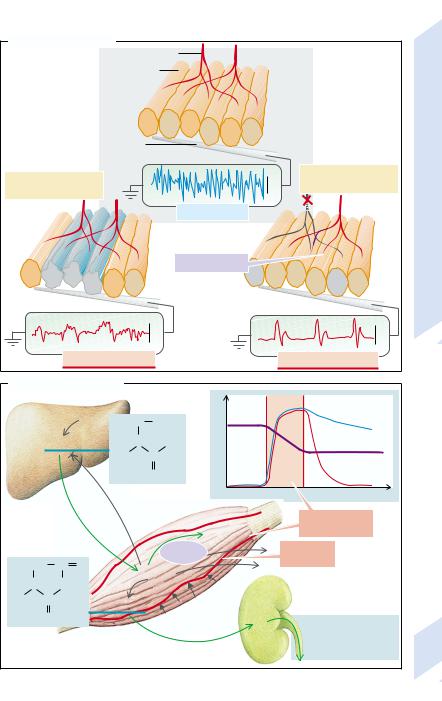
A primary myopathy can be distinguished from a neurogenic myopathy by electromyography (→D).
This is carried out by putting a needle into the muscle and measuring the potential difference from an indifferent electrode on the surface of the skin. As the tip of the intramuscular electrode is largely extracellular, only a fraction of the potential difference across the cell membrane is measured. The amplitude of the recorded changes in potential depends on the number of muscle fibers near the inserted electrode that are depolarized simultaneously.
As all muscle fibers that are innervated by one α-motoneuron are depolarized at the same time, the amplitude of the recorded changes in potential is greater, the higher the density of such fibers is near the electrode. Because the various α-motoneurons are not depolarized simultaneously, the frequency of the changes in potential is a measure of the number of α-motoneurons that innervate the muscle fibers near the electrode.
Normally those muscle fibers in a muscle that are innervated by one α-motoneuron do not lie next to each other, but are distributed over a larger cross-sectional area (→D1). If muscle fibers are destroyed (myogenic myopathy; →D2), the number of muscle fibers near the electrode decreases. This results in a decreased amplitude of the deflection. If an α-mo- toneuron is destroyed (neurogenic myopathy; →D3), the muscle fibers innervated by it do not atrophy evenly, but some of them are taken over by collaterals of neighboring α-moto- neurons. The motor units thus get larger, as does the amplitude of the potential changes. However, the frequency of the deflections is reduced because the muscle fibers near the electrode are now innervated by fewer motor units.
An important pointer to the presence and progression of a muscle disease is provided by the concentrations of creatine, creatinine, and creatine kinase in blood (→E). Creatine is formed in the liver and is eagerly taken up by the intact muscles. Some of the creatine is transformed in the muscles into the anhydride creatinine which, contrary to creatine, easily crosses the cell membrane and is quantitatively excreted by the kidney. The amount of creatinine excreted in the urine per unit of time is thus proportional to the functioning muscle mass. If, as a result of muscular dystrophy, muscle mass is reduced, creatinine excretion decreases (→E1). In acute cell destruction intracellular creatine kinase and creatine are released and their plasma concentrations rise steeply. If there is no further cell destruction, the plasma concentration of creatine kinase drops to normal, but the concentration of creatine may remain elevated, because the creatine formed in the liver is now taken up by fewer muscles. However, creatine production also falls, as it is inhibited by creatine through a feedback mechanism. As a result, plasma concentration or renal excretion of creatine do not parallel the reduction in muscle mass.
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.
D. Electromyography
α-motor neuron
Muscle fiber
|
|
|
|
1 |
|
|
|
|
|
|
|
|
|
Electrode |
|
|
|
|
II |
|
|
|
|
|
|
|
|
and Muscles |
|
Death of individual |
|
1 mV |
Death of individual |
||||||
|
motoneurons |
||||||||
muscle fibers |
|
|
|||||||
|
|
|
|
||||||
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
normal |
|
|
Unit |
|
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
Motor |
|
|
|
|
|
|
Reinnervation |
|
|
|
|
|
|
|
|
|
|
|
10.6 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3 |
|
|
Plate |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
500 V |
|
|
5 mV |
|
|
|
|
Myogenic myopathy |
|
|
Neurogenic myopathy |
|
||
E. Creatine Metabolism |
|
|
|
|
|
||||
|
|
|
|
|
|
1 |
|
Creatine |
|
Liver |
|
|
|
H2C COO– |
Concentration |
|
in plasma |
|
|
|
|
|
N |
NH2 |
|
Creatinine |
|
||
|
Creatine |
|
|
||||||
|
|
in urine |
|
||||||
|
|
|
|
H3C |
C |
|
Creatine |
|
|
|
|
|
|
|
NH2+ |
|
|
kinase |
|
|
|
|
|
|
|
0 |
|
in plasma |
|
|
|
|
|
|
|
|
Time |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Death of |
|
|
|
|
|
|
|
|
Creatine |
muscle cells |
|
|
|
|
|
|
|
|
phosphate |
|
|
|
|
|
|
|
|
Creatine |
Muscle |
|
|
|
CH2 |
C |
O |
|
kinase |
dystrophy |
|
|
||
Creatine |
|
|
|
|
|||||
N |
|
NH |
|
|
|
|
|||
|
|
|
|
|
|
|
|||
CH3 |
C |
|
|
Creatinine |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NH+ |
|
|
|
|
|
Normal excretion: |
|
|
|
|
|
|
Muscle |
|
|
|
||
|
|
|
|
|
|
Creatine |
0 |
|
|
|
|
|
|
|
|
Kidney |
Creatinine 1.8 g/day |
309 |
|
|
|
|
|
|
|
|
|
||
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.

10 Neuromuscular and Sensory Systems
310
Lesions of the Descending Motor Tracts
Spinal α-motoneurons are controlled by several supraspinal neuronal tracts (→A1):
–the pyramidal tract (violet) from the motor cortex;
–the rubrospinal tract from the red nucleus (red);
–the medial reticulospinal tract from the pontine reticular formation (orange);
–the lateral reticulospinal tract from the medullar reticular formation (brown); and
–the vestibulospinal tract (green).
The medial reticulospinal and the vestibulospinal tracts predominantly promote the activity of the so-called antigravity muscles, i.e., the muscles that flex the arms and stretch the legs. The pyramidal, rubrospinal, and lateral reticulospinal tracts, on the other hand, predominantly promote the activity of the flexors of the leg and extensors of the arms.
If the motor cortex or the internal capsule is damaged (e.g., by bleeding or ischemia in the area supplied by the middle cerebral artery), impulse transmission in the immediately adjacent descending cortical tracts is interrupted. These make up the pyramidal tract and other connections of the motor cortex, such as those to the red nucleus and to the medullary reticular formation. The result is a reduced activity not only of the pyramidal tract but also of the rubrospinal and medial reticulospinal tracts. The vestibulospinal and medial reticulospinal tracts are less affected, because they are under stronger noncortical influence, for example, from the cerebellum. An interruption of transmission in the area of the internal capsule thus ultimately results in an excessive activity of the extensors in the legs and the flexors in arms (→A2).
At first, however, spinal shock will set in due to cessation of supraspinal innervation of α- motoneurons (→A3a). The antigravity muscles are also affected, less so than the other muscles though, by the reduced supraspinal activation of the α-motoneurons. In spinal shock the muscles are flaccid and no reflexes are elicited (areflexia).
However, partial “denervation” of the α- and γ-motoneurons as well as of interneurons leads to a gradual increase in sensitivity of these neurons. In addition, the endings of su-
praspinal neurons that are out of action are replaced by synapses with the spinal cord neurons (→A3b). As a consequence, the reflexes gradually gain a stronger influence on the activity of the α-motoneurons, and hyperreflexia occurs.
Another consequence is spasticity. After loss of function of the descending tracts, the activity of the α-motoneurons comes under the increasing influence of the muscle spindles and Golgi tendon organs (→A4). Stretching the muscle spindles stimulates the α-motoneu- rons of the same muscle via a monosynaptic reflex; the increased influence of the muscle spindles results in massive contraction on stretching. Nevertheless, the response of the muscle spindles is mainly phasic, i.e., if they are stretched slowly or continuously their activity slowly decreases. As a result, the influence of the Golgi tendon organs becomes dominant: when the muscle is stretched they inhibit muscle contraction via an inhibiting interneuron. It is also under the influence of the Golgi tendon organs that on slow or continuous stretching the muscle will suddenly become flaccid after initial increase in tone (clasp-knife effect).
The predominance of the stretching muscles leads to extension of the big toe on stroking the sole of the foot (→A5), instead of its normal plantar flexion. This is called Babinski’s sign or the Babinski reflex. It is taken as evidence for a lesion in the pyramidal tract. In fact the Babinski reflex is the result of a lesion of several descending cortical tracts, including the pyramidal tract. Isolated damage of the pyramidal tract (extremely rare) results in neither spasticity nor the Babinski reflex, but only minor disturbance of fine movement.
If the red nucleus has been destroyed (e.g., due to ischemia of the mid-brain or in Wilson’s disease [→p. 252]), coarse tremor will result. Neurons of the red nucleus are, among other functions, important for the dampening of oscillations that can occur as a result of a negative feedback in the control of α-motoneurons. In lesions of the vestibular nucleus abnormalities of balance with vertigo, nystagmus, and nausea predominate (→p. 330).
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.