

книги студ / Color Atlas of Pathophysiology (S Silbernagl et al, Thieme 2000)
.pdf
|
A. Varicosis and Phlebothrombosis |
|
|
|
||
|
1 Normal |
|
|
|
|
|
|
flow conditions |
|
Superficial |
|
Femoralis v. |
|
|
|
|
|
|
|
|
|
Deep |
|
leg vein |
|
Perforating v. |
|
|
leg vein |
|
|
|
(Dodd) |
|
|
|
|
|
Perforating v. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(Hunter) |
|
|
|
|
|
|
Long saphenous v. |
Diseases |
|
|
|
|
|
Popliteal v. |
|
|
|
|
|
|
Perforating v. |
|
|
|
|
|
|
(Boyd) |
|
|
|
|
Flow direction during |
Posterior |
||
|
|
|
Vascular |
|||
|
Venous valves |
|
muscle relaxation |
tibial vv. |
||
|
Perforating v. |
|
(no back-flow on |
|
||
|
|
muscle contraction) |
Perforating v. |
|||
|
Leg muscles |
|
||||
|
|
|
|
(Cockett III) |
||
|
|
|
|
|
Perforating v. |
Peripheral |
|
Genetic risk factors, |
|
Immobilization, |
(Cockett II) |
||
|
|
Perforating v. |
||||
|
standing or sedentary |
coagulopathy, operation, |
(Cockett I) |
|||
|
occupation over years, |
trauma, contraceptive |
|
|||
|
pregnancy, overweight |
pill, tumor |
|
|
||
|
|
|
|
|
|
7.34 |
|
|
|
2 |
3 |
|
Plate |
|
Primary varicosis |
Acute |
|
|||
|
|
|
||||
|
|
|
|
phlebothrombosis, |
|
|
|
|
|
|
risk of embolism |
|
|
|
Venous valve |
|
|
|
|
|
|
failure |
Feeling of heaviness, |
|
|
|
|
|
Flow |
pain, |
|
|
|
|
|
burning sensation |
|
|
|
||
|
reversal |
|
|
|
||
|
and edema in |
|
|
|
||
|
|
|
|
|
||
|
Flow reversal |
the legs |
|
|
|
|
|
on muscle |
|
|
|
|
|
|
contraction |
|
|
|
|
|
|
|
Inflammation |
|
|
|
|
|
|
(varicophlebitis) |
|
|
|
|
|
|
|
|
4 |
|
|
|
|
|
5 |
Pulmonary embol- |
|
|
|
|
|
ism and infarction |
|
|
|
after May, Partsch and Creutzig) |
Chronic venous failure |
|
|
|
||
|
|
6 |
|
|
|
|
part |
|
|
Fibrosis, tissue hypoxia, ulcus |
|
241 |
|
(in |
|
|
|
|||
|
|
|
|
|
|
|
Photo: Siegenthaler W. Differentialdiagnose innerer Krankheiten. 16th Aufl. Stuttgart: Thieme; 1988.
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.

8 |
|
|
|
Metabolism |
S. Silbernagl |
|
|
|
Overview |
Amino Acids |
|
|
|
|
|||
|
|
|
|||
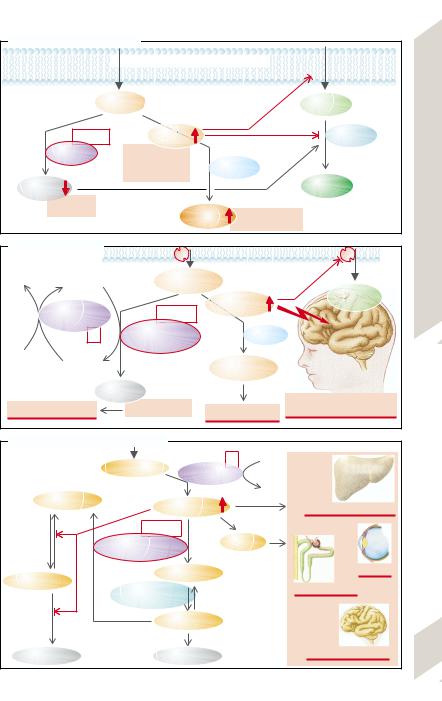
Metabolic abnormalities are frequently caused by faulty endocrine regulation (e.g., diabetes mellitus; →286ff.), or genetic defects of enzymes (enzymopathies) or of transport proteins, the latter, for example, in cystic fibrosis (→p.162) and cystinosis (see below). The endocytosis and exocytosis of lipoproteins can also be affected by defects of apolipoproteins or membrane receptors (→p. 246ff.).
If there is an enzyme defect (→A, enzyme X), the substrate (A) to be metabolized accumulates, so that the concentration of A in the cell organelle, in the cell, and/or in the body rises. This can result in
–substrate A being “stored” and thus becoming a problem, if only in terms of space (storage diseases, e.g., glycogen storage disease, lipidoses; →p. 244);
–toxic effects at high concentrations, or precipitation of the substrate because of its low solubility, in this way causing damage (e.g., cystine in cystinuria or uric acid/urate in gout; →p.120 and 250);
–conversion of the substrate, via another metabolic pathway (enzyme Z), to a metabolite that is harmful at the increased concentration (metabolite E);
–inhibition of the metabolic conversion of another enzyme (enzyme Y) or of a carrier that is essential for the transport of other
substances, too (substrate C).
In addition, the primary enzyme defect leads to a deficiency of the substance formed by this metabolic pathway (→A, metabolite B). In glycogen storage disease, for example, it causes glucose or ATP deficiency (→p. 244). A lack of metabolite B may additionally increase the metabolic rate of other enzyme reactions (→A, enzyme Y).
Metabolic disorders play a part in the processes dealt with in almost every chapter in this book. This chapter describes further examples of metabolic abnormalities, their selection made mainly according to the seriousness, treatability (on early diagnosis), and prevalence of the abnormalities.
242
Amino acids (AA) are both building blocks and breakdown products of proteins. They are the precursors of hormones and transmitters, purines, amines, heme, etc., and they serve as energy sources. Ammonia, produced during catabolism, is incorporated into urea and excreted in this form. Too many or too few amino acids, a carrier defect (→e.g., p. 96ff.), or abnormal formation of urea (→p.174) thus usually lead to marked disorders. A lack of essential amino acids may be due to inadequate intake (unbalanced diet).
In phenylketonuria (PKU) the conversion of phenylalanine (Phe) to tyrosine (Tyr) is blocked (→B1). If as a result the Phe concentration in plasma rises above ca. 1 mmol/L, Phe is broken down via secondary pathways, especially phenylpyruvate that appears in urine (= PKU). In addition, Phe blocks the transport of certain amino acids, so that these neither leave parenchymal cells (sequestration) nor are able to enter brain cells (→B). Severe developmental defects in the brain are the result. A lack of melanin (→B), formed from tyrosine, also disturbs pigmentation (light sensitivity). Early diagnosis and a low-Phe diet can prevent these developmental disorders. Rare forms of PKU are due to a defect of dihydropteridine reductase (→B2).
Further metabolic disorders of amino acids include (the corresponding enzyme defect is given in brackets): hyperglycinemia (propio- nyl-CoA-carboxylase), hyperoxaluria (type I: alanine-glyoxylate aminotransferase; type II: D-glycerate dehydrogenase), maple syrup disease (multi-enzyme complex in the breakdown of branched-chain AA), homocystinuria (type I: cystathionine– β-synthase; type II: methionine resynthesis from homocysteine; →p. 34, A2), cystinosis (carrier defect lysosomal cystine accumulation), alkaptonuria (homogentisic acid dioxygenase), oculocutaneous albinism (phenoloxidase = tyrosinase), and hyperprolinemia (type I: proline dehydrogenase; type II: follow-on enzyme), type I being a partial form of Alport’s syndrome.
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.
A. Effects of Enzyme Defect
Transport 

 Transport
Transport 
Substrate |
|
Substrate |
|
A |
|
C |
|
Defect |
Substrate |
Enzyme Y |
|
A |
|||
|
|
|
Enzyme X |
Storage of A |
|
|
|
|
Acids |
|
|
Toxicity of A |
|
Enzyme Z |
|
||
|
|
Precipitation of A |
|
|
|||
Metabolite |
|
|
|
|
Metabolite |
Amino |
|
|
B |
|
|
|
|
D |
|
|
Deficiency |
|
|
|
|
|
|
|
of B |
Metabolite |
Storage of B |
|
|||
|
|
Overview, |
|||||
|
|
|
E |
|
Toxicity of B |
|
|
B. Phenylketonuria |
|
|
|
|
|
||
|
|
|
|
|
|
|
|
NADP+ |
Tetrahydropteridine |
L-phenylalanine |
|
|
8.1 |
||
|
|
|
|
|
|
Tryptophan, |
Plate |
|
Dihydropteridine |
Defect 1 |
|
L-phenylalanine |
other AA |
||
|
|
|
|
|
|||
|
|
|
|
|
|
||
|
reductase |
|
|
|
|
|
|
|
2 |
Phenylalanine-4- |
|
|
Bypass |
|
|
|
|
monooxygenase |
|
|
|
|
|
NADPH + H+ |
|
|
|
|
|
|
|
|
Dihydropteridine |
|
|
Phenylpyruvate |
|
|
|
|
L-tyrosine |
In urine: |
|
Mental retardation, |
|
||
|
|
Melanin deficiency |
|
|
|||
Light sensitivity |
|
Phenylketonuria |
neurological disorders |
|
|||
C. Galactosemia (see next page for text) |
|
|
|
|
|
||
|
Galactose |
2 |
ATP |
|
|
Galactokinase |
|
|
|
|
|
|
|
|
|
|
|
ADP |
|
Glucose-1-P |
Galactose-1-P |
Hepatomegaly, jaundice |
|
|
|
|
|||
|
Defect 1 |
|
|
|
|
Galactose-1-uri- |
Galactitol |
|
|
|
dyltransferase |
|
|
|
|
UDP-galactose |
Cataract |
|
|
Glucose-6-P |
|
|
||
UDP-galactose- |
Damaged tubules |
|
||
|
|
|||
|
4-epimerase |
|
|
|
|
UDP-glucose |
|
|
|
Glucose |
|
Glycogen |
Mental retardation |
243 |
|
|
|
||
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.

Carbohydratis
|
Abnormalities of carbohydrate metabolism are |
||
|
usually caused by enzymopathies or abnormal |
||
|
regulation (see also anemia, →p. 30ff. or dia- |
||
|
betes mellitus, →p. 286ff.). |
|
|
|
In galactosemia (→p. 243 C) galactose is |
||
|
split off from lactose in the gut and can be chan- |
||
|
ged into glucose or glycogen, especially in the |
||
|
liver. In case of a galactose-1-uridyl transferase |
||
|
deficiency |
(→p. 243 C1), |
galactose-1-phos- |
|
phateaccumulates in many organs with the on- |
||
|
set of breast-feeding. The organs are damaged |
||
|
as galactose-1-phosphate inhibits enzymes |
||
|
which are active in glucose metabolism. Dam- |
||
|
age can also be caused by galactitol, formed |
||
|
from galactose-1-phosphate. Early diagnosis |
||
Metabolism |
and a galactose-freediet canpreventsuch dam- |
||
hypergalactosuria is less serious. |
|||
|
age (uridine diphosphate galactose can still be |
||
|
formed). A galactokinase deficiency (→p. 243 |
||
|
C2) associated with hypergalactosemia and |
||
8 |
In hereditary fructose |
intolerance (→A, |
|
|
center) there is a defect of fructose-1-phos- |
||
|
phate aldolase. The breakdown of fructose |
||
|
(fruits, saccharose) is blocked and fructose-1- |
||
|
phosphate accumulates. This inhibits phos- |
||
|
phorylase and fructose-1,6-diphosphate aldo- |
||
|
lase in the liver, thus causing hepatogenic hy- |
||
|
poglycemia, |
acute liver failure, or cirrhosis |
|
(→p.172ff.). If diagnosed early and the patient put on a fructose-free diet, life expectancy is normal, while fructose infusion can quickly be fatal due to liver failure.
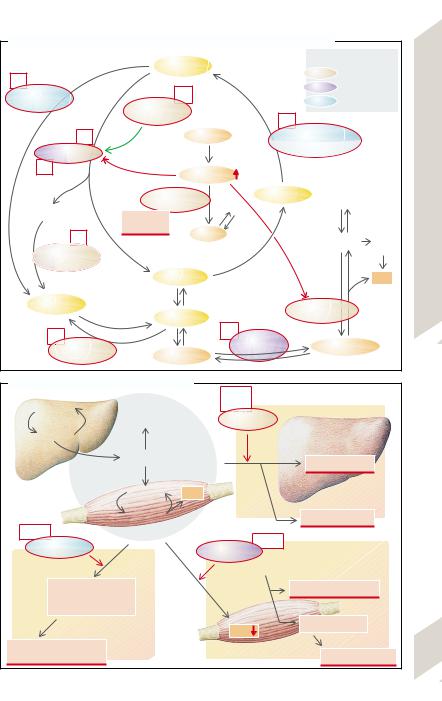
Glycogen storage diseases. Glucose is stored in muscles and liver as glycogen. Breaking it down provides glucose that is used locally or reaches other organs (→A,B). If the breakdown of glycogen is blocked, glycogen overloading and hypoglycemia result. This is caused by enzyme deficiencies.
Several types are distinguished (→A): type Ia (von Gierke; glucose-6-phosphatase deficiency); type Ib (deficiency of microsomal glucose-6-phosphate translocase [not shown in diagram]); type II (Pompe; lysosomal α-glucosidase deficiency); type III (Forbes, Cori; debrancher enzyme deficiency, the most common type); type V (McArdle; muscle phosphorylase deficiency); type VI (Hers; hepatic phosphorylase deficiency); and type VIII (Huijing; hepatic phosphorylase b kinase deficiency). A (very rare) deficiency of
244 glycogen synthesis (type IV; Andersen; brancher enzyme deficiency) results in glycogenosis because an
abnormal type of glycogen is stored in the brain, heart, muscle, and liver. In type VII (Tauri; muscle phosphofructokinase deficiency), on the other hand, glucose is prevented from being utilized to provide the muscles with energy.
Depending on the primary effects of the enzyme deficiencies, one can simplify the classificationbydividingtheglycogenstoragediseases intolivertypes(I,III,VI,VIII),muscletypes(V,VII), and other types (II, IV) (→B). In the liver types hepatomegaly (due to excess deposition of glycogen)andhypoglycemiaaretheprominentfeatures, while in the muscle types it is largely energy deficiency. Physical work does not increase plasma lactate and leads to rapid fatigue, muscle cramps and muscle pain as well as to myoglobinuria (in type V), which may cause renal failure. The effects of type II (cardiomegaly, weakness of respiratory muscles) and type IV (liver failure) often end in death in childhood.
Lipidoses
Lipidoses are disorders of fat metabolism, in which defects of enzymes and other proteins cause the accumulation (and thus deposition) of lipids.
In Gaucher’s disease there is a lysosomal β-glucocer- ebrosidase (β-glucosidase) deficiency, in which glucocerebroside accumulates (adult form) in the spleen, liver, lung, and bone marrow (Gaucher cells), hypersplenism (thrombocytopenia), spontaneous fractures as well as pneumonia and cor pulmonale being some of the consequences. In Niemann–Pick disease (five phenotypes, A–E) there is an accumulation of sphingomyelin and cholesterol in the lysosomes. In types A (80% of all cases of the disease) and B there is a deficiency of sphingomyelinase, while in type C1 the deficiency is of a protein (NPC1) which plays an important role in the intracellular distribution of cholesterol. The effects of type A are enlargement of several organs and severe neurological abnormalities that can be fatal already in childhood. A deficiency of acid lipase is the cause of cholesterol-ester storage disease (liver cirrhosis and atherosclerosis) and in Wolman’s disease (the infantile form of acid lipase deficiency). The gangliosidoses (e.g., Tay–Sachs and Sandhoff’s disease) are caused by various defects of the hexosaminidases and their activators, or of β-galactosidase. In most forms the accumulated gangliosides lead to very severe cerebral disorders and death in early childhood. In Refsum’s disease the breakdown of phytanic acid is blocked (defect of phytanic acid-α-hydroxylase), as a result of which it accumulates and, incorporated into myelin, leads to polyneuropathy.
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.
A. Causes of Glycogen Storage Diseases I–VIII and Fructose Intolerance
Lysosomal breakdown |
Glycogen |
|
|
|
Enzyme defects |
|
|
|||
|
|
|
|
|
Enzymes |
esp. in liver |
|
|||
II |
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
Enzymes |
esp. in skeletal |
|
||
Lysosomal |
|
|
VIII |
|
|
|
|
|||
|
|
|
|
|
|
muscles |
|
|||
α-glucosidase |
Phosphorylase- |
|
|
|
Enzymes |
in many organs |
|
|||
|
|
|
|
|
|
|
|
|||
|
kinase |
|
|
|
IV |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
VI |
|
|
Fructose |
|
Branching enzyme + |
|
|
|||
Phosphorylase |
|
|
|
|
Glycogensynthetase |
|
Lipidoses |
|||
|
|
|
|
|
|
|
|
|
||
V |
|
|
Fructose-1-P |
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
Fructose- |
|
UDP-glucose |
Lactate |
|
Carbohydratis, |
|||
Limit dextrin |
|
1-P-aldolase |
|
|
|
|
||||
|
|
|
|
|
|
|
||||
Fructose |
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|||
III |
intolerance |
Trioses |
|
|
|
|
|
Citrate |
||
|
|
|
|
|
Pyruvate |
|||||
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
cycle |
|||
Debranching |
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
||
enzyme |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Glucose-1-P |
|
|
|
|
|
ATP |
Plate8.2 |
|
Glucose |
|
Glucose-6-P |
|
|
Fructose-1,6-P2- |
|
||||
|
|
|
|
|
aldolase |
|
|
|||
Ia |
|
|
VII |
Phospho- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
Glucose-6- |
|
|
|
|
fructokinase |
|
Fructose-1,6-P2 |
|
||
phosphatase |
|
Fructose-6-P |
|
|
|
|
|
|
|
|
B. Effects of Glycogen Storage Diseases |
I, III, |
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
||
Glycogen |
Other |
VI, VIII |
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|||
|
organs |
|
Liver types |
|
|
|
|
|
||
Glucose |
|
|
|
|
|
|
|
|
|
|
Liver |
Blood glucose |
|
|
|
|
Hepatomegaly |
|
|
||
|
|
|
|
|
|
|
|
|
||
|
Glucose |
ATP |
|
|
|
|
|
|
|
|
Muscle |
Glycogen |
|
|
|
|
Hypoglycemia |
|
|
||
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
||
II, IV |
|
|
|
|
V, VII |
|
|
|
|
|
Other types |
|
|
|
|
|
|
|
|
|
|
|
|
Muscle types |
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
During severe muscle activity: |
|
|
|||
Cardiomegaly, |
|
|
|
|
|
Cramps, weakness |
|
|
||
muscle weakness |
|
|
|
|
|
|
|
|
|
|
(respiratory muscles!) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ATP |
Myoglobinuria |
|
|
|||
|
|
|
|
|
|
|
|
|
||
Cardiorespiratory failure |
|
|
|
|
|
|
|
Renal failure |
245 |
|
|
|
|
|
|
|
|
|
|
||
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.

8 Metabolism
Abnormalities of Lipoprotein Metabolism
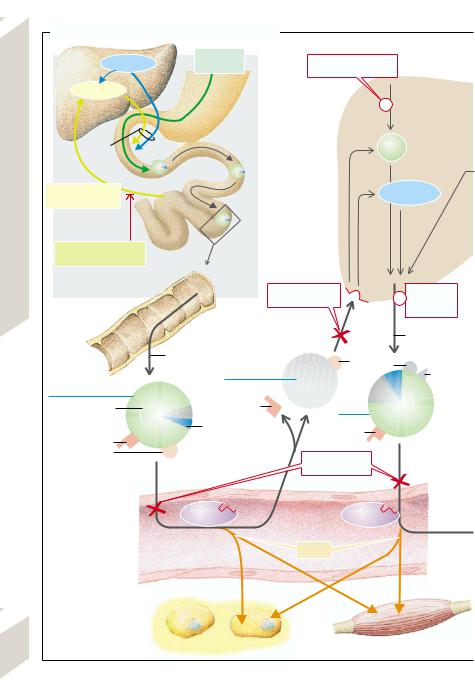
Among the disorders of fat metabolism there are, in addition to the lipidoses (→p. 244), mainly those diseases in which the concentrations of lipoproteins in serum and thus lipid transport in blood are abnormal. Lipids are transported in blood in globular molecular complexes (microemulsions), the lipoproteins (LPs). Their surface consists largely of amphiphilic lipids (phospholipids and nonesterified cholesterol), while their “core” contains nonpolar (hydrophobic) lipids, i.e., triglycerides (TGs) and cholesterol ester (Chol-E), the transport and storage form of cholesterol. The LPs also contain certain apolipoproteins (Apos). The LPs differ in the size, density (which gives them their name, see below), lipid composition, site of origin as well as their apo(lipo)proteins (see Table), the latter serving as structural elements of the LP (e.g., ApoAII and ApoB48), as ligands (e.g., ApoB100 and ApoE) for LP receptors in the membrane of the LP target cells, and as enzyme activators (e.g., ApoAI, ApoCII).
The chylomicrons transport lipids from the gut (via the gut lymphatics) to the periphery (skeletal musculature, fat tissue), where their ApoCII activates the endothelial lipoprotein lipase (LPL); thus free fatty acids (FFAs) are split off which are taken up by the cells of the muscles and fat tissue (→A2). In the liver the chylomicron remnants bind to receptors (LDL re- ceptor–related protein [LRP]?) (→A9) via ApoE, they are endocytosed and in this way deliver their TGs as well as their cholesterol and cholesterol esters. Such imported as well as newly synthesized TG and cholesterol are exported by the liver (→A4) in very low density LP (VLDL) to the periphery, where they activate LPL with their ApoCII, also leading to the release of fatty acids (→A3). ApoCII is lost in
this process and ApoE is exposed. This leaves VLDL remnants or intermediate density LP
(IDL), half of which return to the liver (binding mostly with ApoE to the LDL receptors). They are freshly loaded with lipids in the liver, leaving the liver as VLDL (→A4). The other half of the IDL is transformed (with loss of ApoE and exposure of ApoB100) on contact with hepatic lipase to low density LP (LDL). Two thirds of these LDLs deliver their cholesterol and Chol- E to the liver (→A7), one third to extrahepatic tissues (→A14), both processes requiring the binding of ApoB100 to the LDL receptors. By binding to receptors, mediated by clathrin in the coated pit regions of the cell surface, LDLs undergo endocytosis in which the LDL receptors recirculate to the cell membrane. After fusion of the endosomes with lysosomes, the apolipoproteins are “digested” and the Chol-E split, so that free cholesterol reaches the cytosol (→A5). As a result of this rise in the concentration of intracellular cholesterol: 1) the key enzyme of cholesterol synthesis is inhibited (3-HMG-CoA reductase); 2) cholesterol is again esterified to its storage form (activation of acyl-CoA-cholesterol-acyl transferase [ACAT]); and 3) LDL receptor synthesis is inhibited.
The high density LPs (HDLs) exchange certain apolipoproteins with chylomicrons and VLDLs and also take up excess cholesterol from extrahepatic cells (→A10) and blood. By means of their ApoAI they activate the plasma enzyme lecithin-cholesterol acyltransferase ([LCAT] which in part esterifies the cholesterol) and pass on cholesterol and Chol-E to the liver, among other organs, and to those steroid hormone–producing glands (ovaries, testicles, adrenals) which have HDL receptors (→A6).
|
|
Lipoprotein- |
|
% of |
Apolipoproteins |
Formations in |
Transport function |
|
|
class* |
TG |
ChoL |
|
or [from] |
|
|
Chylomicr. |
90 |
3 |
AI, B48, CII + III, E |
Gut |
TG etc.: Gut Periphery |
|
|
Chyl. remnants |
|
|
|
[Chylomicr.] |
Lipids: Gut Liver |
|
|
VLDL |
65 |
15 |
B100, CII + III, E |
Liver |
TG etc.: Liver Periphery |
|
|
|
IDL |
|
|
B100, CIII, E |
[VLDL,HDL] |
Lipids: Liver, LDL |
|
|
LDL |
10 |
45 |
B100 |
[IDL] |
Cholesterol: IDL Liver, Periphery |
246 |
|
HDL |
5 |
20 |
AI,III + IV, CIII, D |
Periphery |
Cholesterol: Periphery IDL |
|
|
|
|
|
|
|
|
* Electrophoretic separation distinguishes between α-lipoproteins (= HDL), pre-β-lipoproteins (= VLDL) and β-lipoproteins (= LDL).
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.

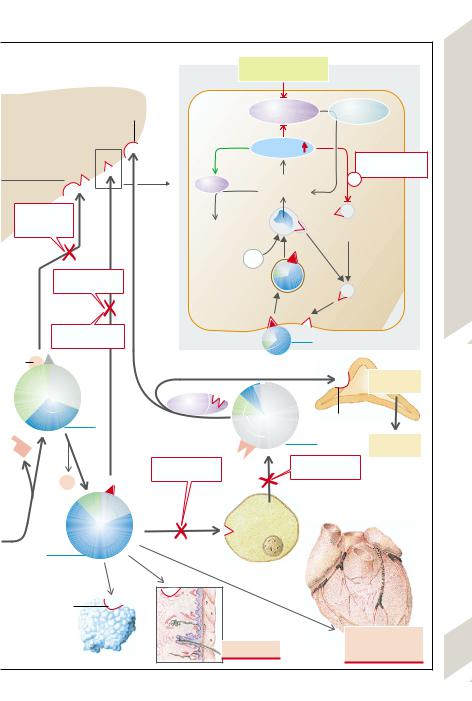
An increase in blood lipids can affect cholesterol, triglycerides or both (hypercholesterolemia, hypertriglyceridemia or combined hyperlipidemia). Hyperlipoproteinemia is currently the all-inclusive term.
With most patients who have hypercholesterolemia (> 200– 220 mg/dL serum) there is an increased familial prevalence of the condition, but the cause remains unknown (polygenic hypercholesterolemia). However, overweight and diet play an important role. LDLcholesterol can be lowered most importantly by a preference in the diet for vegetable (unsaturated) fats. Animal (saturated) fats, on the other hand, raise cholesterol synthesis in the liver and in consequence lower its LDL receptor density (→A7) so that the concentration of cholesterol-rich LDL in serum is raised (LDL-cholesterol > 135 mg/dL). As a result, there is an increased binding of LDL to the scavenger receptor that mediates the incorporation of cholesterol in macrophages, skin, and vessel walls (→A8). Hypercholesterolemia is thus a risk factor for atherosclerosis
(→ p. 236ff.) and coronary heart disease (→ p. 218).
In familial hypercholesterolemia (hyperlipoproteinemia type IIa; incidence of homozygotes is 1:106; of heterozygotes 1:500) the plasma cholesterol is markedly raised from birth (twice as high in heterozygotes; six times as high in homozygotes) so that myocardial infarction may occur even in children. The primary causes are defects in the gene for the high-affinity LDL receptor which prevents the cellular uptake of LDL (→A7,14). The defect can cause: 1) diminished transcription of the receptor; 2) receptor proteins remaining in the endoplasmic reticulum; 3) a reduced incorporation of the receptor into the cell membrane; 4) reduced LDL binding; or 5) abnormal endocytosis. Serum cholesterol rises as a result, firstly, of a reduction in the cellular uptake of cholesterol-rich LDL and, secondly, of extrahepatic tissues synthesizing more cholesterol, because the reduced LDL uptake in these tissues fails to inhibit the action of 3-HMG- CoA reductase (→A5). Treatment consists, in addition to an appropriate diet (see above), of administering ionic exchange resins (cholestyramine) that bind bile salts in the gut and thus prevent their enterohepatic recirculation
(→A1). This increases the fresh synthesis of bile salts from cholesterol in the liver and thus reduces the intracellular cholesterol concentration. In heterozygotes this increases the LDL receptor density (→A5). However, it also stimulates cholesterol synthesis, but this in turn can be prevented by administering inhibitors of 3-HMG-CoA reductase (e.g., lovastatin) (→A5). The treatment of homozygotes includes the removal of LDL from plasma by plasmapheresis.
In another single-gene defect, combined hyperlipidemia (hyperlipoproteinemia type IIb), the TGs as well as cholesterol are slightly raised. The cause is possibly an overproduction of ApoB, so that an increased synthesis of VLDL occurs (→A4) and therefore more LDL is also formed. Familial dys-β-lipoproteinemia predisposes to hyperlipoproteinemia type III. In this condition, instead of the normal ApoE3, an ApoE2 variant is expressed that is not recognized by the E receptor. As a result, the hepatic uptake of chylomicron remnants and of IDL is disturbed (→A9,13), so that their plasma concentration rises (high risk of atherosclerosis;
→p. 236ff.).
Primary hypertriglyceridemia is due to in-
creased TG synthesis in the liver (→A11) or (rarely) to abnormalities in the breakdown of chylomicrons and VLDL (hyperlipoproteinemia type I), the result of a deficiency of LPL or ApoCII (→A2,3). They predispose a person to, for example, pancreatitis (→p.158ff.); in addition HDLs are reduced and thus the atherosclerosis risk is increased (reduced removal of cholesterol from the vessel wall?).
Gene defects can also result in subnormal LP concentrations (hypolipoproteinemia). Familial hypo-α-lipoproteinemia (Tangier disease) is due to a defect of ApoA and there is a HDL deficiency (→A10), increasing the atherosclerosis risk. In A-β-lipoproteinemia there are no LDLs in plasma (hypocholesterolemia). This is caused by an abnormal synthesis of ApoB, so that chylomicrons cannot be exported from the gut mucosa, nor can VLDL from the liver. This produces accumulation of TG in both organs.
Lipoprotein Metabolism
247
!
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.
|
A. Lipoprotein Metabolism and its Abnormalities |
|
|
|
|||
|
Liver |
Cholesterol |
|
Food fats |
Primary |
|
|
|
|
|
hypertriglyceridemia |
|
|||
|
|
|
|
|
|||
|
Bile salts |
|
|
|
+ TG synthesis |
||
|
|
|
|
|
11 |
||
|
|
Bile |
|
|
|
TG |
Liver |
|
Enterohepatic |
|
|
|
Cholesterol |
||
|
circulation |
|
|
|
|
|
|
8 Metabolism |
Bile salt-binding |
|
|
|
|
|
|
|
ion exchanger |
|
|
|
|
|
|
1 |
|
|
|
Familial dys-β- |
|
Combined |
|
|
|
|
|
|
|||
|
|
|
|
lipoproteinemia |
+ hyper- |
||
|
|
|
|
|
9 |
4 |
lipidemia |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
HDL contact |
|
|
|
HDL contact |
|
|
in blood |
|
|
|
|
E |
|
|
||
|
|
|
in blood |
Chylomicron |
(E) |
|
|
|
|
|
|
remnants |
|
|
(B100) |
|
Chylomicrons |
|
|
|
|
|
|
|
Triglycerides (TG) |
|
CII |
VLDL |
TG |
|
|
|
Apolipo- |
|
Cholesterol (Chol) |
|
|
||
|
|
|
|
|
|||
|
proteins: |
|
CII |
|
|
||
|
+ chol. ester (Chol-E) |
|
|
||||
|
|
CII |
12 |
|
|
||
|
|
|
|
|
|
||
|
|
E |
|
|
Primary hyper- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
triglyceridemia |
|
|
|
|
|
|
|
Capillary endothelium |
|
|
|
|
2 |
|
LPL |
LPL |
|
|
|
|
|
|
|
3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
FFA |
|
|
248 |
|
|
|
Fat cells |
Musculature |
|
|
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.
|
|
|
|
|
3-HMG-CoA- |
|
|
|
|
|
|
|
5 |
|
reductase inhibitor |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3-HMG- |
Cholesterol |
II |
||
|
|
|
|
|
I + |
||||
|
|
HDL receptor |
Liver cell |
CoA-reductase |
synthesis |
||||
|
|
|
|
|
|
|
Metabolism |
||
|
|
|
|
|
Cholesterol |
|
|
||
|
|
|
|
|
|
|
|
Polygenic hyper- |
|
|
|
|
|
|
|
|
+ |
cholesterolemia |
|
|
|
|
|
|
Free |
|
|
||
|
|
|
ACAT |
|
|
|
Lipoprotein |
||
|
|
7 |
|
|
cholesterol |
|
|
||
|
|
|
|
|
|
|
|
||
|
Familial |
|
|
|
|
|
|
|
|
|
dys-β-lipo- |
13 |
Chol-E |
|
|
|
Receptor |
||
|
proteinemia |
|
|
|
|
||||
|
|
|
|
Lysosome |
|
synthesis |
|||
|
|
|
|
|
|
|
|
+ 8.4 |
|
|
|
|
|
|
|
|
|
|
|
|
Polygenic hyper- |
|
|
|
|
|
|
8.3 |
|
|
cholesterolemia |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
Plate |
||
|
|
|
|
Endocytosis |
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
Familial hyper- |
LDL receptor |
|
|
|
|
|||
|
|
|
|
LDL |
|
|
|
||
|
cholesterolemia |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
||
E |
(B100) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
6 |
|
|
|
|
Endocrine |
|
|
|
|
|
|
|
|
|
glands |
|
|
|
|
LCAT |
|
|
|
|
|
|
|
|
IDL |
|
|
|
|
HDL receptor |
|
|
|
|
|
|
A I |
HDL |
|
Steroid |
|
|
CII |
|
|
|
|
|
||||
|
|
|
|
hormones |
|
||||
|
|
|
|
|
|
|
|||
|
|
|
Familial hyper- |
|
10 |
Hypo-α- |
|
|
|
|
E |
|
cholesterolemia |
|
lipoproteinemia |
|
|
||
|
B100 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
14 |
|
LDL |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Chol |
|
|
receptor |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
LDL |
+ Chol-E |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
8 |
|
|
Extrahepatic |
|
|
|
|
|
|
|
|
cells |
|
|
|
|
|
|
Scavenger |
|
|
|
|
|
|
|
|
|
receptor |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Atherosclerosis, |
|
|
|
|
|
Skin |
|
Xanthoma |
|
esp. coronary |
249 |
|
|
|
Macrophage |
|
|
heart disease |
||||
|
|
|
|
|
|
|
|
|
|
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.

8 Metabolism
250
Gout
Gout is the result of chronically elevated uric acid/urate concentration in plasma (hyperuricemia: > 7 mg/dL).
Uric acid formation. Uric acid (UA) is the end-product of purine metabolism (→A1). However, normally 90% of the resulting nucleotide metabolites adenine, guanine, and hypoxanthine are reused in that they are reformed to AMP, IMP, and GMP by adenine phosphoribosyltransferase (APRT) and hypoxanthine guanine phosphoribosyltransferase (HGPRT), respectively. Only the remainder is converted to xanthine and further to uric acid by xanthine oxidase (XO) (→A1). The low solubility of urate and especially of uric acid, which decreases even further in the cold and at low pH (pKa′ of urate/uric acid ≈5.4), is the reason why gout develops from hyperuricema.
The renal excretion of uric acid (→A2) is ca. 10% of the filtered amount, i.e., the UA/urate concentration in the final urine is 10– 20 times higher than in plasma. Drugs with uricosuric activity (e.g., benzbromarone) can increase UA/urate excretion and thus lower their plasma concentration.
Hyperuricemia occurs in ca. 10% of the population in western industrialized countries; one in 20 develops gout (men > women). 90% of patients with the condition have primary gout (→A3) with a genetic disposition. The underlying primary hyperuricemia is due to the fact that the renal excretion of UA can match normal UA production only when the UA concentration in plasma, and thus in the glomerular filtrate, is raised (asymptomatic hyperuricemia). If there is a higher purine intake
(especially in innards, meat extract, fish, mussels, etc.), this is even more the case, and thus in the long term sodium urate crystals are precipitated again and again. On rare occasions the hyperuricemia is caused by a partial lack of HGPRT, in which case the proportion of reutilized nucleotide metabolites (see above) falls, and thus more UA is formed (→A1). (In the Lesch–Nyhan syndrome there is a complete absence of HGPRT. In this disease childhood gout is paralleled by severe central nervous system abnormalities.)
As the solubility of urate is especially low in synovial fluid and at low temperature, and
since the digits are cooler than the body core, urate crystals are often formed in the acral joints of the foot (microtophi). Alcohol, which increases adenine nucleotide metabolism, favors crystal deposition as does obesity, certain drugs (e.g., diuretics), and a high lead load. The often increased urinary concentration of UA/ urate in hyperuricemia results in the formation of urinary stones (→A5 and p.120).
An attack of gout (→A4) occurs when the urate crystals (possibly as a result of trauma) are suddenly released from the microtophi and are recognized by the immune system as foreign bodies. An aseptic inflammation of the joint develops (arthritis, →A4; see also p. 48ff.), attracting neutrophils which phagocytize the urate crystals. When the neutrophils subsequently break down, the phagocytized urate crystals are released again, which maintains the process. A very painful, deep-red joint swelling occurs, in 70– 90% of first attacks affecting one of the proximal toe joints.
Acute urate nephropathies (→A5). If the UA concentration in plasma and primary urine suddenly rises markedly (usually in secondary gout; see below) and/or (because of low fluid intake), the urine is highly concentrated and the urine pH low (e.g., in protein-rich diet), large amounts of UA/urate may be precipitated in the collecting duct with plugging of the lumen. Acute renal failure may result (→p.108).
Repeated attacks of gout (chronic gout) can damage the joints (also hands, knees, etc.) to such an extent that, under constant pain, marked joint deformities with destruction of cartilage and bone atrophy will occur (→A4, photograph). There may also be circumscribed deposits of urates (tophi) around the joint or at the edge of the auricles as well as in the kidneys (chronic gouty nephropathy).
So-called secondary hyperuricemia or gout is initiated by, for example, leukemia, tumor treatment (raised nucleotide metabolism) or by renal failure with other causes (reduced UA excretion).
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.