

2.2. Регуляція клітинного циклу. Апоптоз. Онкогенетика.
Мітотічеський цикл і його регуляція. Роль циклинов і циклинзависимых киназ. Принципи передачі митогенного сигналу. Роль чинників зростання, интегринов і кадгеринов. Контрольні точки митотического циклу. Апоптоз.
Генетичні механізми канцерогенезу. Загальна характеристика генів, що беруть участь в канцерогенезі: вірусні онкогены, протоонкогены, гены-супрессоры пухлин, гени-мутатори. Канцерогенні чинники.
Мітотічеський цикл і його регуляція. Здавалося б, давно відома річ – фази циклу (G1, S, G2, M), стадії митоза (профаза, метафаза, анафаза і телофаза), - все вже сто років як описано і вивчено.
Але що примушує клітку «рухатися» по цьому кругу? Як вдається їй забезпечити строго впорядковану зміну безлічі подій, складових клітинний цикл? Які механізми запускають, наприклад, синтез ДНК в S-фазе, а потім під час його зупиняють, попереджаючи повторне подвоєння ДНК? Як і чому відбувається руйнування ядерної оболонки в пізніше профазе, а потім освіта відразу два таких оболонок в телофазе? І так далі – подібні питання доречні відносно кожного процесу.
Довгий час все це було досконало неясно. І лише завдяки дослідженням останніх років перед нашим поглядом почала проступати, з одного боку, надзвичайно дивовижна (по своїй складності і «продуманості»), а з іншого боку, цілком природна картина під назвою «Регуляція клітинного циклу».
Впродовж клітинного циклу різні клітки можуть вступати в різні процеси.
Дійсно, клітки, що діляться, можуть не тільки продовжувати ділитися або тимчасово припинити ділення, впавши в «сплячку» (як буває із стволовими клітинами). Їх доля, залежно від цілого ряду обставин (генетичної програми, дії гистогормонов, вплив інших внутрішніх і зовнішніх чинників), може дуже круто змінитися. Ось найбільш драматичні варіанти:
- клітка вступає в процес диференціювання
- у клітці запускаються механізми самознищення (апоптоза)
- клітка піддається бласттрансформации, тобто перетворюється на пухлинну клітку.
Все це процеси надзвичайної біологічної важливості. І те, в який саме з них вступить клітка, що ділиться, визначається в ході клітинного циклу.
Тривалість клітинного циклу змінюється залежно від типу клітки і стадії розвитку організму. Наприклад, клітинний цикл заплідненого яйця дуже нетривалий. Запліднене яйце жаби ділиться дуже швидко (близько 30 хвилин). Велику частину цього часу займають S і М фази, а на фази G1 і G2 витрачається дуже мало часу. Оскільки в яйці вже існує великий запас білків і інших речовин, необхідних для клітинного ділення, синтезу нових білків майже не вимагається. Таким чином, G1,2- фази коротшають.
На ранніх стадіях розвитку необхідно сформувати якомога більше кліток відносно короткий проміжок часу. Наприклад, в заплідненому яйці жаби протягом 6 годин відбувається 12 дроблень і утворюється ембріон з 8192 кліток. Клітини діляться швидко, але збільшуються в розмірах трохи. Проте, за рахунок величезної кількості кліток і деякій ассиметрии ділення, ембріон формується протягом декількох годинників. Після того, як ембріон сформований, і закладка трьох основних типів тканини завершена, час, необхідний для клітинного циклу, збільшується за рахунок подовження G-фаз. Синтез нових білків приводить до того, що клітка збільшується в розмірі і об'ємі.
Клітинний цикл заплідненого яйця людини, на відміну від яйця жаби, триває значно довше. Перше ділення триває 30 годин, а весь процес дроблення – 7 діб (у жаби – 6 годин).
Проте зразкова тривалість стадій циклу для кліток людини, що швидко діляться, складає:
S-период – 10 ч
G2-период – 4,5 ч
М-період – 0,5 ч
G1-период – 9 ч.
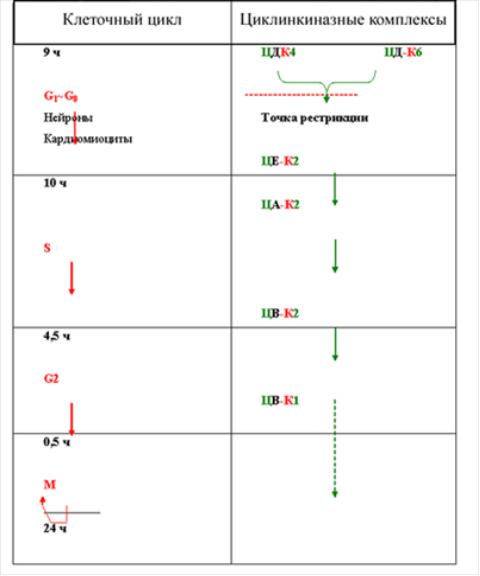
Разом – 24 ч (рис.42.). Але, зрозуміло, це дуже приблизні оцінки, і для якихось кліток цикл може бути тривалішим. Зокрема наголошувалося, що для сперматогоний S-период триває 15 годин. І, відповідно, в 1,5 разу більше опиняється біля них тривалість циклу – приблизно 1,5 діб. Отже на 10 митотических ділень сперматогоний (що відбуваються на першому етапі сперматогенезу) потрібний 2 тижні.

Ріс.42. Клітинний цикл: митоз і цитокинез (клітинне ділення) складають М-фазу циклу, кульмінацією, якою є утворення двох дочірніх кліток. Кожна дочірня клітка вступає в G1-период интерфазы і може почати новий клітинний цикл. За періодом G1 слідує S-фаза, під час якої ДНК і хромосоми дуплицируются, і далі – фаза G2. Початок митоза означає кінець интерфазы. Клітки, що покояться, затримуються у фазі G1 і, як то кажуть, знаходяться у фазі G0. Зазвичай эукариотические клітки, які не зупинилися у фазі G0, завершують цикл за 24 ч.
Роль циклинов і циклинзависимых киназ. Дослідження митотического циклу показали, що ключову роль в почерговій зміні його фаз грають спеціальні протеинкиназы - тобто циклинзависимые киназы (Cdks - cyclin-dependent kinasis). Кожна з них фосфорилирует певні білки, залучені у відповідну фазу циклу, і таким чином активує або інгібірує їх.
Молекула будь-якої Cdk складається лише з однієї субъединицы, яка сама по собі неактивна. Для активації ж Cdk потрібне пов'язання з нею спеціального білка - циклина (Ц). Є декілька разных циклинов, і, як вважають, що зв'язався з Cdk циклин не тільки активує фермент, але і додає йому субстратную специфічність відносно тих або інших білків.
Термін циклин відображає той факт, що концентрації циклинов в клітці протягом клітинного циклу змінюються циклічним чином (рис.43) .
У активній формі протеинкиназы є гетеродимерные комплексами циклин-Cdk (Ц-cdk), де циклин служить активаторной, а Cdk - каталітичною субъединицей. Дані утворення не розглядають як єдині молекули, а називають комплексами, через те, що можуть бути різні поєднання конкретних циклинов і конкретних Cdk, причому кожне таке поєднання характерне для строго певної фази циклу.
Запускають цикл комплекси циклинD-Cdk4 і (або) циклинD-Cdk6. Разниє циклины позначаються латинськими буквами, а разные Cdk - арабськими цифрами. Названі комплекси функціонують на початковій стадії постмитотического (G1) періоду і, викликаючи відповідні внутріклітинні події, сприяють переходу кліткою «точки рестрикції» (крапка, передуюча S-периоду, тобто для кліток, що постійно діляться, знаходиться десь в кінці G1-периода, а для клітки, початкуючої ділення після перерви, десь в кінці Go -периода). Аналогічно, ті ж комплекси приводять до повернення «сплячої» Go -клетки в митотический цикл.
Друга половина G1 -происходит під впливом комплексу циклин E-Cdk2, що управляє.
У наступному - синтетичному (S) - періоді функціонуюча циклинзависимая киназа залишається тій же (Cdk2), але вона двічі міняє своїх циклиновых партнерів - ними послідовно стають циклин А і циклин Ст. Відповідно, міняються і ті білкові субстрати, на які діють Cdk2.
У премитотическом (G2) періоді останній з вищеназваних циклинов (В) зв'язується з іншою Cdk - Cdkl. Саме комплекс циклин B-Cdkl «вводить» клітку в митоз і «керує» цим складним процесом. Тому його ще називають митоз-стимулирующим чинником - MPF (від mitosis-promoting factor).
З складу останніх двох комплексів виходить, що їх субстратная специфічність визначається не тільки циклином, але і самій Cdk, а найвірніше – одночасно тим і іншим, тобто наявним поєднанням циклина з Cdk.
Cdk1 нерідко позначається інакше - Cdk2. Річ у тому, що мутації гена Cdkl, порушуючи клітинний цикл, викликають у дріжджів фенотип виду Сdc2 (при якому клітки стають незвичайно довгими). Тому, ще до ідентифікації функції цього гена, він був позначений як Сdc2.
Ріс.43. Схема динаміки клітинного циклу
Отже, вся різноманітність подій клітинного циклу управляється відносно невеликим числом комплексів циклинов і Cdk.
Способи регуляції і активності Cdks. Зважаючи на виняткову функцію циклинзависимых киназ (Cdks), їх зміст і особливо активність знаходиться під складним контролем. Ось принципові способи цього контролю.
1. Регуляція синтезу Cdks. Мабуть, не все Cdks одночасно присутні в клітці на різних стадіях її циклу. Тому дуже важливим моментом є активація гена тій або інший Cdk.
Так, відомо, що комплекси G1 періоду (циклин D-Cdk4,6 і циклин E-Cdk2), крім інших численних дій, запускають транскрипцію гена киназы Cdkl. Остання ж необхідна для утворення комплексу G2- періоду і митоза (циклин B-Cdkl або MPF). Таким чином, про синтез компонентів даного комплексу клітка «піклується» завчасно, ще до реплікації ДНК.
2. Регуляція активності Cdks. Способи цієї регуляції достатньо різноманітні.
а) Один з них - пов'язання з якою-небудь Cdk активаторной субъединицы - циклина.
б) Другий спосіб - скріплення (або тільки з Cdk, або зі всім комплексом циклин-Cdk) ингибиторной субъединицы. В ролі останньої виступають спеціальні білки (р15, р16, р21, р27, р57). Відомо, принаймні, два сімейства таких білків. Білки сімейства INK4 (р15 і р16) зв'язуються з Cdk4,6 і тим самим перешкоджають утворенню комплексів (циклин D-Cdk4,6), що запускають клітинний цикл. Білки другого сімейства, KIP1 (р21,р27, р57), зв'язуються з вже сформованими комплексами - і це теж приводить до ефекту, що інгібірує.
с) Третій спосіб контролю активності Cdks - це їх фосфорилування і дефосфорилирование. Тобто дані протеинкиназы (Cdks) самі здатні регулюватися тим же самим способом, яким вони управляють активністю «підвідомчих» ним білків.
Фосфорилування по одних локусам Cdk володіє активуючою дією; фосфорилування інших ділянок, навпаки, інгібірує фермент.
Але активність киназ відновлюється під дією специфічної фосфатази, що кодується геном cdc25a. З позначення виходить, що це ще один ген, мутація якого у дріжджів приводить до порушення клітинного циклу і фенотипу cdc.
Виявлені також дві тирозинкиназы (ТК), що інгібірують іншу Cdk, - Cdkl; одна з цих ТК, мабуть, діє на вільну Cdkl, а інша - на весь комплекс циклин В -Cdkl.
3.Регуляція синтезу і розпаду активаторів і інгібіторів Cdks. Як приклад звернемося спочатку до циклинам.
а) Багато митогенные чинників, «спонукаючи» клітку до ділення, запускають такі регуляторні ланцюги, які, врешті-решт, активують ген циклина D (крім багатьох інших дій). На наступних стадіях клітинного циклу стимулюється синтез чергових циклинов і так далі
б) Украй интересным є також управління розпадом циклинов. Воно здійснюється за допомогою убиквитин залежного механізму, який використовується для короткоживучих білків.
Найчіткіше роль цього механізму показана для циклина В, комплексу циклин, що бере участь в освіті, В - Cdk2 і митоз-стимулирующего чинника (MPF, він же комплекс циклин B-Cdkl). Фосфоріліруя певні білки, MPF стимулює входження клітки в митоз. Але для завершення митоза необхідні багато в чому прямо протилежні події. Тобто зміст MPF повинен знижуватися. Це досягається за допомогою швидкого розпаду циклина Ст.
Зразкова послідовність подій. Максимальної активності комплекс MPF досягає в метафазі митоза. В цей час він фосфорилирует, крім інших білків, так званий чинник АРС (аnaphase-promotoring complex) тобто чинник, що забезпечує анафазу. А даний чинник є нічим іншим, як убиквитинлигазой, специфічною відносно MPF. Тому він починає послідовно приєднувати (одну за іншою) молекули убиквитина до циклину В. Меченний таким чином циклин В швидко руйнується в протеосомах. У результаті зміст комплексу циклин B-Cdkl значно знижується, і в клітці благополучно здійснюються події анафазы, а потім і телофазы. Потім в G1-периоде чинник АРС инактивируется, від чого швидкість розпаду циклина В знижується, і даний циклин починає накопичуватися в клітці.
в) Контроль за синтезом речовин, Cdks, що інгібірують (або комплекси циклин-Cdk). Регуляція синтезу часто використовується позаклітинними эффекторами для впливу на клітинний цикл - стимуляції або гальмування проліферації. Один з прикладів: у деяких регуляторних внутріклітинних ланцюжках фігурують білки сімейства Smad. Ці білки, утворюючи відповідні чинники транскрипцій, стимулюють синтез інгібіторів р15, р21 і ін. У результаті активність комплексів G1-периода гальмується і клітка припиняє ділення.
Принципи передачі митогенного сигналу. Практично всі сигнальні шляхи, регулюючі проліферацію кліток, «націлені» на комплекси G1-периода — в основному, циклин D—Cdk4,6 і, у меншій мірі, циклин Е—cdk2. Це і зрозуміло: як вже наголошувалося, саме дані комплекси «запускають» черговий клітинний цикл.
Розглянемо два митогенных сигнальних шляху. Один з них відноситься до стволових клітин епітелію і починається з скріплення ЕФР — епідермального чинника зростання.
Другий шлях активується в Т-хелперах після їх взаємодії з АПК — антигенпредставляющими клітками.
Тепер на цих же двох прикладах покажемо «вихід» подібних шляхів на комплекси циклин-Cdk.
У обох випадках зовнішній сигнал приводить до активації тирозинкиназы, що асоціюється з рецептором. Це веде (через ті або інші посередники) до запуску каскадів митогенактивируемых протеинкиназ (МАПК). Кінцеві ферменти даного каскаду, фосфорилируя ряд чинників (Elk, Ets, ATF2, Tcf і ін.) транскрипцій, активують їх, а через них — і т.з. гени ранньої відповіді (FOS, JUN). У культурі кліток вже через 30 мін після початку дії митогена активність цих генів досягає максимального рівня.
Продукти сімейств генів FOS і JUN — це знову-таки чинники транскрипцій, але специфічні вже відносно інших генів — т.з. генів сповільненої відповіді. Тому через деякий час починається експресія цих генів.
Серед останніх-то і знаходяться наші «знайомі» — гени циклина D, Cdk4 і Cdk6, тобто компонентів комплексів циклин-Cdk, специфічних для першої половини G1 - періоду циклу.
Крім
того, активуються ще деякі гени і серед
них — ген білка Мус.

 Після свого синтезу білок Мус, у свою
чергу, впливає на активність ряду генів.
Так, він гальмує експресію гена білка
р27
— інгібітору цілого ряду комплексів
циклин-
Cdk.
І одночасно білок Мус активує ген Cdc25a,
який кодує специфічну фосфатазу. Остання
дефосфорилирует
Cdk4
і Cdk2,
що приводить до їх активації.
Після свого синтезу білок Мус, у свою
чергу, впливає на активність ряду генів.
Так, він гальмує експресію гена білка
р27
— інгібітору цілого ряду комплексів
циклин-
Cdk.
І одночасно білок Мус активує ген Cdc25a,
який кодує специфічну фосфатазу. Остання
дефосфорилирует
Cdk4
і Cdk2,
що приводить до їх активації.
В результаті ми отримуємо наступні ефекти:
збільшення вмісту в клітці циклина D і реагуючих з ним киназ — Cdk4, Cdk6;
зниження змісту ряду інгібіторів Cdks;
підвищення активності (в результаті дефосфорилирования) тих же циклинзависимых киназ (Cdk4, Cdk6), а також Cdk2.
Все це і забезпечує формування в клітці достатньої кількості активних комплексів циклин D—Cdk4,6, початківців готувати клітку до ділення.
Відмітимо, щоб розглянуті сигнальні шляхи «спрацювали», необхідна одна умова: клітка повинна бути фіксована на якому-небудь позаклітинному матриксі.
Дія антимитогенов. Це дія ФНО (чинника некрозу пухлин) на пухлинні клітки. ФНО «запускає» в клітках відразу декілька сигнальних шляхів, і серед них один містить ферментативный каскад МАПК. Тільки тепер збудження відповідного рецептора через ряд посередників (серед яких — сфингозин і ПК-С) приводить до гальмування МАПК. Далі — все як в попередніх шляхах, але з протилежним знаком. Отже, у результаті в клітці різко знижуватиметься кількість активних комплексів циклин D—Cdk4,6, внаслідок чого ділення припиняться. Дія ж ФНО через інші сигнальні шляхи ініціюватиме апоптоз.
Другий приклад: мова йде про Тфрр — трансформуючому чиннику зростання. Вже наголошувалося, що він пригноблює проліферацію багатьох кліток. Рецептори Тфрр, мабуть, схожі по структурі з рецепторами ЕФР (епідермального чинника зростання). Це означає, що при скріпленні свого лиганда рецепторні субъединицы об'єднуються в димерные структури, домени цитоплазми кожної субъединицы володіють тирозинкиназной активністю, в активованому стані ці домени фосфорилируют один одного. Відмінність полягає лише в тому, що в даному випадку модифіковані домени рецептора зв'язують інший комплекс білків цитоплазми: тепер це комплекс Smad2+Smad3. Він теж фосфорилируется рецепторною тирозинкиназой, після чого набуває здатності зв'язувати третій білок — Smad4.
Потім весь цей потрійний комплекс диффундирует в ядро і виконує роль чинника транскрипції для генів, що кодують інгібітори Cdks. Маються на увазі такі білки, як р15 і р21.
Накопичення в клітці цих інгібіторів і приводить до гальмування проліферації.
Роль чинників зростання, интегринов і кадгеринов. Кліткам нижчих організмів для вступу до S-фазу клітинного циклу необхідна наявність живильних речовин в зовнішньому середовищі. При недоліку живильних речовин клітина не ділиться. У вищих організмів наявність живильних речовин зазвичай не є лімітуючим чинником. Проте сигнал, що ініціює ділення клітки, частіше буває зовнішнім, чим внутрішнім. Доказу цього були отримані в перших експериментах з культурою кліток ссавців in vitro.
При розробці методики вирощування кліток ссавців в культурі було відмічено, що клітки ростуть краще, якщо вони знаходяться усередині кров'яних згустків. Із-за незручності дослідження кліток, що ростуть, усередині згустка, після згортання крові згусток віддаляється. Рідина, що залишилася, відома як сироватка. Якщо видалити всі клітки до того, як кров згорнеться, і додати отриману сироватку до кліток, що ростуть, вона не підтримуватиме зростання. Це доводить, що при згортанні з кліток крові вивільняється якийсь чинник, необхідний для ділення клітин. Було показано, що ця речовина вивільняється з тромбоцитів, і воно було назване тромбоцитарным чинником зростання (PDGF). Виявляється, нормальні клітки ссавців містять достатню кількість живильних речовин для ділення. Таким чином, в клітки повинен поступати додатковий сигнал, стимулюючий проліферацію. У випадку з тромбоцитарным чинником зростання цей білок секретируется тромбоцитами при згортанні крові після утворенні рани. Для заповнення тканинного дефекту тромбоцитарный чинник зростання, один з головних чинників зростання організму, сигналізує навколишнім кліткам про необхідність приступити до ділення.
До теперішнього часу виділено і охарактеризовано велике число чинників зростання: тромбоцитариый чинник зростання; епідермальний чинник зростання (EGF); чинники зростання фибробластов (FGF) (мають дев'ять изоформ і володіють невеликою клітинною специфічністю, проте ще відносяться до групи малоспецифічних); чинник зростання нервів (NGF) (цей чинник зростання діє тільки на клітки нервової системи); эритропоэтин (ЕРО) (цей чинник зростання стимулює утворення еритроцитів в кістковому мозку); иитерлейкин-2 і інтерлейкін-3.
Всі ці чинники зростання діють в основному на специфічні клітки-мішені. Кожен чинник зростання є лигандом, який зв'язується із специфічним поверхневим рецептором клітки і ініціює процес передачі сигналу.
Більшість кліток ссавців знаходяться в особливій фазі клітинного циклу, званою Go-фазой або компартментом клітинного диференціювання. Ця фаза клітинного циклу є подовженою фазою G1. Це означає, що гени, що кодують білки, які запускають проліферацію кліток, знаходяться в «вимкненому» стані. У цьому положенні метаболічна енергія клітки витрачається на утворення спеціалізованих білків, необхідних для здійснення диференціювання. Таким чином, гени, що кодують ці білки, знаходяться в «включеному» стані.
У більшості типів кліток гени для клітинної проліферації можуть бути включені знову. Проте в деяких клітках гени проліферації ніколи не запускаються повторно після виключення. При завершенні проліферації в клітках серцевого м'яза Cdk і циклины вимикаються і більше ніколи не включаються. То ж справедливо і для всіх типів нейронів.
Проте у відповідних умовах багато кліток можуть повернутися в цикл ділення. Практично у всіх тканинах організму відбуваються клітинні «перетворення». Зазвичай такі перетворення відбуваються рідко. Виняток становлять клітки крові і кишкового епітелію, в яких швидкість оновлення кліток дуже висока.
Як показано вище, один з основних типів сигналів, активуючих Cdk і циклины, генерують чинники зростання. Проте, навіть отримавши такий сигнал, організм повинен регулювати відповідь так, щоб підтримувати кількість і просторову організацію кліток. Якщо цього не відбувається, наступають летальні зміни організму.
При дослідженні нормальних кліток, вирощених в культурі тканини, було показано, що існують різні чинники, що визначають число кліток, необхідних для підтримки архітектури тканини, і якість з'єднання кліток з базальною мембраною. Якщо вирощувані в культурі фибробласты заповнюють всю чашку Петрі, вони перестають ділитися і, очевидно, йдуть в псевдо-Со-фазу. Якщо в стерильних умовах по поверхні культури провести шпателем діагональну лінію, клітки, розташовані на цій лінії, гинуть і залишається бесклеточная діагональ. Клітки, що знаходяться на краю «рани», починають негайно ділитися і пролиферировать, заповнюючи вільний простір. Як тільки простір заповнений, ділення знову припиняється.
На основі цього спостереження був зроблений вивід про те, що нормальні клітини діляться до досягнення певної щільності. Після утворення контактів з іншими клітками на субстраті, всередину клітки передається сигнал про те, що всі контактні крапки зайняті, і гени, що запускають проліферацію, вимикаються.
Якщо клітці не давати стикнутися з поверхнею тарілки, вона зберігає округлу форму і не вступає в митоз. Якщо ж клітка стикається з поверхнею і розплющується, вона формує з поверхнею тарілки контакти, звані фокальними контактами і фокальними адгезионными пластинками. До складу фокальної адгезионной пластинки входять важливі компоненти цитоскелета, включаючи актиновые филаменты, мікротрубочки і інші структурні білки. Через деякий час ці клітки вступають в митоз.
Результати цих і аналогічних експериментів доводять, що молекули навколишнього середовища, що наприклад входять до складу міжклітинних контактів і контактів кліток з міжклітинною речовиною, беруть участь в управлінні клітинною проліферацією.
Тепер розглянемо такі сигнальні шляхи, які починаються від адгезивних мембранних білків. Одні з цих шляхів стимулюють проліферацію кліток; інші, навпаки, гальмують.
Багато кліток здатні ділитися, тільки будучи прикріпленими до позаклітинної структури – базальної мембрани (эпителиоциты), колагеновим волокнам (фибробласты) і так далі
Інформація ж про зв'язок клітки з такою структурою поступає від интегринов. Це адгезивні білки з двома нерівними субъединицами.
При пов'язанні з позаклітинним матриксом менша (р-) субъединица интегрина активує одну з тирозинкиназ — FAK (Focal Adhesion Kinase), а та, у свою чергу, ще одну тирозинкиназу — Src. Остання відноситься до класу нерецепторних тирозинкиназ, оскільки не входить до складу рецепторного комплексу і навіть не взаємодіє безпосередньо з рецепторами.
Безпосереднім субстратом Src є білок SHC, який пов'язує даний сигнальний шлях (що йде від интегринов) з сигнальними шляхами від рецепторів митогенов. Загальною ж частиною останніх доріг є багато разів згадувані вище каскади МАПК (митогенактивируемых протеинкиназ). Отже, адаптерний білок SHC повинен стимулювати проходження сигналу по даних каскадах.
Проте не цілком ясно, який саме член каскадів активується білком SHC. Є припущення, що це той же комплекс білків (GRB, SOS, Ras, Raf), що і у разі дії ЕФР (епідермального чинника зростання).
Але, на наш погляд, треба врахувати наступну обставину. Для того, щоб клітка ссавців почала ділитися, необхідне одночасне виконання двох умов: прикріплення клітки до якої-небудь поверхні і дія на неї ростового чинника.
Причому в неприкріпленій клітці проходження сигналу від ростового чинника блокується на одній з МАПК, а конкретно — на киназе МЕК (Мек1).
Звідси витікає, що:
а) сигнал від интегринов йде саме до цієї ланки МАПК — киназе МЕК;
б) дана киназа активується тільки при дії на неї відразу двох білків: попередньої киназы з сімейства МАПК і білка SHC (після дії на нього Src).
Тоді і виходить, що сигнали від ростового чинника і интегринов взаємно доповнюють один одного, а кожен з них окремо не здатний індукувати ділення клітки.
Ефект прикріплення клітки до опори включає ще одну важливу подію. Воно стосується «знаменитого» чинника транскрипції — білка р53.
Останній активує гени, що зупиняють ділення (зокрема, ген білка р21 — інгібітору комплексів циклин- Cdk), а також гени апоптоза. Тому недивно, що що йде від интегринов сигнал призводить до зниження вмісту в клітці такого «шкідливого» білка.
У разі білка р53 зазвичай регулюється не синтез, а розпад. Тому під дією сигналу від интегринов, мабуть, тим або іншим способом прискорюється протеолиз цього білка. За деякими даними, це може бути пов'язано із зниженням змісту іншого білка (ARF), що інгібірує розпад білка р53.
Контактне гальмування проліферації. Якщо ж клітка встановлює контакт не з позаклітинним матриксом, а з іншими клітками, то спостерігається ефект, прямо протилежний попередньому, — припинення ділень. Це позначається як контактне гальмування.
Після чергового пересівання кліток в плоску скляну судину («матрац») вони спочатку — протягом декількох годинників прикріпляються до дна судини і лише після цього входять в клітинний цикл (стимуляція ділень).
Коли ж на дні судини не залишається вільного місця і клітки вступають в контакт один з одним, ділення припиняються (контактне гальмування).
Сигнал про міжклітинний контакт, мабуть, йде від кадгеринов — адгезивних білків, що беруть участь в утворенні таких контактів. Ймовірно, у кадгеринов міняється при цьому конфігурація; тому їх домени цитоплазми набувають здатності зв'язувати білок р-катенин.
Припускають, що даний білок — чинник транскрипції. Коли він вільний від зв'язку з кадгерином (тобто коли клітка ще не контактує з іншими клітками), він утворює активний комплекс з ще одним чинником транскрипції — білком Tcf4. Комплекс мігрує в ядро і тут прямим або (швидше за все) опосередкованим способом стимулює транскрипцію генів циклина D і білка Мус.
Це знов ті самі гени, які активуються в раніше розглянутих сигнальних шляхах, — що йдуть від ростових чинників і від интегринов. Ті шляхи сходяться і взаємно доповнюють один одного на каскадах митогенактивируемых киназ (МАПК).
Але шлях, в-катенин, що містить, теж є необхідним для ділення. При уловлюванні в-катенина кадгерином ділення припиняються. Отже, цей шлях також необхідно доповнює попередні. На наш погляд, все пояснюється і встає на свої місця при наступних припущеннях:
на початку будь-якого клітинного циклу повинен утворитися якийсь (завжди один і той же) промитотический комплекс чинників (ПМКТФ) транскрипцій, специфічний відносно генів ранньої відповіді;
до складу цього комплексу входять в-катенин і білок Tcf4, що можливо лише тоді, коли в-катенин не зв'язаний кадгерином (немає міжклітинних контактів);
крім того, комплекс містить чинники, що активуються каскадом МАПК; причому цей каскад сам, по попередньому нашому припущенню, вимагає подвійного сигналу від интегринов (клітка прикріплена до позаклітинної структури) і від ростового чинника.
У результаті число умов, необхідних для ділення клітки (в принципі здібною до ділення), зростає вже до трьох: це не тільки прикріплення до поверхні і дія ростового чинника, але і відсутність контактів з іншими клітками.
Лише при виконанні цих умов активуються гени ранньої відповіді, і запускається механізм клітинного циклу, зокрема, утворюється «друга черга» чинників транскрипцій, а потім і продукти стимульованих ними генів — циклин D, Cdk4,6 і так далі
Але цим перипетії внутріклітинних регуляторних шляхів, пов'язані з в-катенином, не вичерпуються. Встановлено ще, принаймні, два интересных факту.
а) Деградація в-катенина проводиться за допомогою убиквитинзависимого механізму. Причому як убиквитинлигазы (ферменту, переносячого убиквитин на Р-катенін) виступає вже знайомий нам чинник АРС, що забезпечує анафазу.
Останній виконує таку ж функцію відносно циклинов, необхідних для входження клітки в цикл. Тому немає нічого дивовижного, що одночасно з даними циклинами руйнується і чинник транскрипції, потрібний для їх синтезу. Відбувається ж це в метафазі і анафазе митоза, коли АРС активується шляхом фосфорилування.
За деякими даними, перед взаємодією з АРС в-катенин теж повинен піддатися фосфорилуванню — киназой-ЗР-гликогенсинтетазы (GSK-3P), яка, у свою чергу, знаходиться під контролем ряду білків.
Така «підвищена увага» з боку клітки до в-катенину — непряме свідоцтво його ключовій ролі в запуску ділення. Це цілком узгоджується з припущенням, що в-катенин — необхідний компонент універсального комплексу транскрипції, початківця клітинний цикл при будь-якому митогенном стимулі.
б) Другий факт полягає в тому, що при контактному гальмуванні в клітці збільшується зміст білка р53. Це теж виглядає цілком природно, якщо вважати, що все той же комплекс чинників транскрипцій, крім іншого, прискорює розпад білка р53, вимикаючи ген білка ARF — інгібітору протеолиза білка р53.
Звичайно, відомо тут ще далеко не все. Але про принцип роботи і деякі деталі цього чудового механізму вже можна сказати.
Що стосується принципу роботи, то він достатньо простий і очевидний. Комплекс циклин-Cdk чергової стадії циклу, як правило, повинен забезпечити три процеси:
а) «виведення з гри» комплексу попередньої стадії
б) стимуляцію подій «своєї» стадії
в) утворення (або активацію) комплексу наступної стадії.
Виходить свого роду «ланцюговий» механізм, після включення, якого кожна стадія процесу готує умови для переходу до наступної стадії. Поза сумнівом, такий же принцип багатьох інших складних біологічних процесів — таких, як ембріональний розвиток, диференціювання, регенерація.
Подивимося тепер, як конкретно реалізується цей принцип у разі клітинного циклу.
Контрольні точки митотического циклу. (Білок р53, мікротрубочки: зв'язок з клінікою). Точна реплікація і розподіл генетичного матеріалу — це найважливіші умови виживання клітки. У клітинному циклі існують чотири крапки, в яких точність реплікації, правильність послідовності і рівне розділення ДНК контролюються спеціальними клітинними механізмами.
A. Контрольна точка фази G1. Якщо у фазі G1 виявляється пошкодження ДНК, білок р53 виступає в ролі чинника транскрипції і викликає затримку кліток в G1. Нестабільний р53 зазвичай швидко руйнується. Проте, коли в клітці з'являється аномальна ДНК, білок р53 стабілізується і приєднується до цієї ДНК. В результаті р53 накопичується в ядрі і стимулює експресію білка, Cdk2, що інгібірує.
Клітка затримується у фазі G1 до тих пір, поки пошкоджені нуклеотиды не будуть відновлені ферментами репарації. Затримка у фазі G1 запобігає копіюванню пошкоджених підстав і гальмує мутацію ДНК. При відщеплюванні білка р53 від ДНК його концентрація знижується; інгібітор Cdk відділяється, і Cdk починає экспрессироваться.
У клітках багатьох метастатичних пухлин людини обидва аллеля р53 неактивні. У таких клітках порушений нормальний контроль, здійснюваний р53, і пошкоджена ДНК може реплицироваться. В деяких випадках це приводить до метастатичної трансформації пухлин.
Б. Контрольная точка S-фазы функціонує у фазі S, коли реплицируется ДНК. Поява мутацій в процесі реплікації і їх подальше вбудовування в геном може викликати серйозні наслідки, включаючи загибель клітки. Якщо відбулися помилки в реплікації, (що трапляється) і якщо вони були пропущені репаративными ферментами, клітка не може вийти з S-фазы. Перевірка точної реплікації ДНК — найважливіша регуляторна точка клітки.
B. Контрольна точка С2-фази. Нерепліцированная ДНК блокує перехід клітки від С2 -фазы до М-фази. Відбувається катастрофічне пошкодження, якщо клітка проходить через фазу циклу, незавершив молекулярні процеси, необхідні для підготовки до ділення. Наприклад, при введенні MPF в клітки, що знаходяться в S-фазе, неповністю реплицированные хромосоми конденсуються, а потім фрагментуються.
Г. Контрольная точка М-фази. Причиною зупинки циклу в даній крапці може бути неправильна збірка веретена ділення. Наприклад, неприкріплення кинетохоры какой- або хроматиды до мікротрубочок веретена ділення. Мікротрубочки (МКТ) постійно полімеризуються і деполимеризуются. Під час митоза відбуваються різкі і непередбачувані переходи МКТ з того, що росте (полімеризація) в стан, що скорочується (деполимеризация), - це катастрофа і назад – це порятунок. Медикаментозна терапія онкологічних хворих полягає в руйнуванні митотического веретена колхицином, винбластином і винкристином, які порушують полімеризацію МКТ.
Залежно від результатів «перевірки», вибирається один з трьох варіантів:
а) безупинний перехід до наступної стадії циклу;
б) затримка на поточній стадії для виправлення виявлених дефектів;
в) запуск механізму апоптоза, якщо виявлені порушення непоправні.
У більшості, якщо не у всіх, випадках хромосомних пошкоджень центральну роль в зупинці циклу грає білок р53.
Роль білка р53 («Вартовий генома», «Диспетчер апоптоза», Пухлинний супрессор). Білок р53 контролює виключно важливі клітинні процеси і, завдяки цьому, залучений у велику кількість всіляких регуляторних ланцюгів. Він (або його ген) активується у відповідь на різноманітні пошкодження клітинної структури: нерепарированные розриви і інші пошкодження ДНК, порушення розбіжності хромосом в митозе, руйнування мікротрубочок і так далі
Сам же білок р53 регулює активність, принаймні, трьох груп генів:
1) активує гени (Р21, GADD45 та інші), що відповідають за зупинку клітинного ділення;
2) активує гени (BAX, KILLER/DR5, PIG та інші), запускаючі апоптоз – процес, ведучий, шляхом активації спеціальних ферментів, до загибелі клітки; а так само репресує гени (BCL2, RELA), стримуючі апоптоз;
3) активує гени (TSP1, BAI1 та інші), гальмуючі ангиогенез (утворення нових судин).
У результаті через посередництво білка р53 клітка у відповідь на пошкодження своєї структури
- або затримується на тій або іншій стадії митотического циклу і виправляє ці пошкодження;
- або (при неможливості виправлень) взагалі припиняє ділення і вступає в процес клітинного старіння (фаза III по Хейфліку);
- або (при потенційній небезпеці пошкодженої клітки для її оточення) здійснює апоптоз, т. е., просто кажучи, самогубство.
Зокрема, апоптозу, крім інших, піддаються і клітки, в яких відбулася пухлинна трансформація. В зв'язку з цим зрозуміло, чому одночасно гальмується ангиогенез: це ще один спосіб обмеження пухлинного зростання.
Тому білок р53 – один з найбільш важливих пухлинних супрессоров. У більшості пухлин функції білка р53, що вже розвиваються, опиняються в тому або іншому відношенні порушені.
Така загальна біологічна роль білка р53, а зараз розглянемо деякі пов'язані з ним питання детальніше.
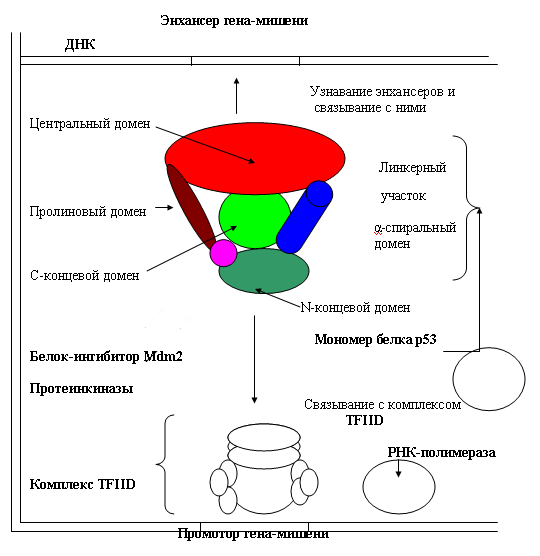
Білок р53 складається з 392 амінокислот, створюючих 6 доменів (мал. 44):
1) N-концевой домен активує ген білка р21 (інгібітор киназ), а так само інших білків, що зупиняють ділення клітки.
2) додатковий домен транскрипції для активації генів-мішеней.
3) гнучкий пролиновый домен проявляє супрессорную активність, запускає апоптоз.
4) центральний домен зв'язується з энхансерами. У цій ділянці мутації 5-8 экзонов приводять до онкогенезу.
5) б-спиральный домен відповідає за ядерну локалізацію р53 і утворення тетрамеров (мономери не активні).
6) С-концевой домен – мішень для модифікуючих ферментів (киназ, ацетилаз, гликозилаз).


Ріс.44. Білок р53
Якщо С-концевой домен не модифікований, центральний домен не здатний взаємодіяти з ДНК-мішенню. Модифікація ж С-домена не тільки додає білку р53 таку здатність, але і впливає на його специфічність. Річ у тому, що р53-зависимые энхансеры (що відносяться до різних генів) декілька розрізняються послідовністю нуклеотидных пар. І від виду модифікації С-домена залежить, з якими конкретно энхансерами зв'язуватиметься білок р53, а з якими – ні.
Таким чином, за допомогою модифікації двох кінцевих ( N- і С-) доменів білок р53 отримує (від дуже численних «джерел») інформацію про стан клітки, переробляє її шляхом зміни своїй конфігурації і належним чином реагує як чинник транскрипції певних генів.
С-концевой домен виконує ще одну функцію. Як наголошувалося вище, деякі гени (BCL2, RELA) білком р53 не активуються, а репресуються. Це дія, як вважають, здійснюється С-доменом. При цьому останній (замість N-домена) зв'язується з комплексом TFIID і пригнічує його активність.
Апоптоз (Програмована загибель кліток). У Англії в Кембріджському університеті на початку 60х років Сідней Бреннер вивчав нематоду Сaenorzhabditis elegans – 1мм довжиною, прозора (видно ділення клітин). Співробітник його лабораторії Джон Салстон описав схему розвитку з яйцеклітини дорослого черв'яка. При розвитку нематоди утворюється 1090 кліток, з яких 131 клітка йде в апоптоз, залишається 959 кліток. Гени смерті, що приводять клітки до апоптозу, встановив американець Роберт Хорвіц. Згодом, завдяки цим роботам, вони стали лауреатами Нобелівської премії в області молекулярної біології.
Термін «апоптоз» з'явився в науці в 1972г. I. Kerr запозичував його у Гіппократа, в перекладі з грецького «апоптоз» – осінній листопад.
Апоптоз - програмована клітинна загибель, енергетично залежний, генетично контрольований процес, який запускається специфічними сигналами і позбавляє організм від ослаблених, непотрібних або пошкоджених кліток. У організмі здорової людини клітинний гомеостаз визначається балансом між загибеллю і проліферацією кліток. Щодня, приблизно близько 5% кліток організму піддаються апоптозу, а їх місце займають нові клітки. В процесі апоптоза клітка зникає безслідно протягом 15-120 хвилин. Апоптоз – це біохімічно специфічний тип загибелі клітки, який характеризується активацією нелизосомных ендогенних эндонуклеаз, які розщеплюють ядерну ДНК на маленькі фрагменти. Морфологічно апоптоз виявляється загибеллю одиничних, безладно розташованих кліток, що супроводжується формуванням округлих, оточених мембраною тілець (“апоптотические тільця”), які тут же фагоцитируются навколишніми клітками.
Це енергозалежний процес, за допомогою якого віддаляються небажані і дефектні клітки організму. Він грає велику роль в морфогенезі і є механізмом постійного контролю розмірів органів. При зниженні апоптоза відбувається накопичення кліток, приклад – пухлинне зростання. При збільшенні апоптоза спостерігається прогресивне зменшення кількості кліток в тканині, приклад – атрофія.
Призначення апоптоза в клітинних популяціях можна сформулювати таким чином:
підтримка чисельності кліток в популяції на заданому рівні;
визначення цього рівня і його зміна під впливом зовнішніх (по відношенню до клітки) сигналів аж до повної елімінації даного типу кліток;
селекція різновидів кліток усередині популяції (зокрема елімінація кліток з генетичними дефектами).
Морфологічні прояви апоптоза. Апоптоз має свої відмітні морфологічні ознаки, як на светооптическом, так і на ультраструктурном рівні. При забарвленні гематоксилином і эозином апоптоз визначається в одиничних клітках або невеликих групах кліток. Апоптотічеськие клітки виглядають як округлі або овальні скупчення інтенсивно эозинофильной цитоплазми з щільними фрагментами ядерного хроматина. Оскільки стиснення клітки і формування апоптотических тілець відбувається швидко і також швидко вони фагоцитируются, розпадаються або викидаються в просвіт органу, то на гістологічних препаратах він виявляється у випадках його значної вираженості. До того ж апоптоз – на відміну від некрозу – ніколи не супроводжується запальною реакцією, що також утрудняє його гістологічне виявлення.
Апоптоз – це механізм загибелі кліток, який має ряд біохімічних і морфологічних відмінностей від некрозу (рис.45). Найчіткіше морфологічні ознаки виявляються при електронній мікроскопії.
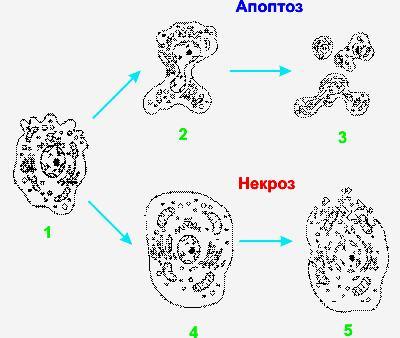
Ріс.45. Зміна ультраструктури кліток тварин при некрозі і апоптозе: 1 – нормальна клітка, 2 – апоптотическое зморщення клітки з утворенням пузирчастих выростов, 3 – фрагментація клітки з утворенням апоптотических везикул, 4 – набухання клітки при некрозі, 5 – некротична дезинтеграція клітки.
Для клітки, апоптозу, що піддається, характерне її стиснення і конденсація хроматина.
Стиснення клітки. Клітка зменшується в розмірах; цитоплазма ущільнюється; органеллы, які виглядають відносно нормальними, розташовуються компактніше. Передбачається, що порушення форми і об'єму клітки відбувається в результаті активації в апоптотических клітках трансглютаминазы. Цей фермент викликає прогресивне утворення перехресних зв'язків в білках цитоплазми, що приводить до формування своєрідної оболонки під клітинною мембраною, подібно ороговевающим кліткам епітелію.
Конденсація хроматина. Це найбільш характерний прояв апоптоза. Хроматін конденсується по периферії, під мембраною ядра, при цьому утворюються чітко обкреслені щільні маси різної форми і розмірів. Ядро ж може розриватися на два або декілька фрагментів. Механізм конденсації хроматина вивчений досить добре. Він обумовлений розщеплюванням ядерної ДНК в місцях, що зв'язують окремі нуклеосомы, що приводить до розвитку великої кількості фрагментів, в яких число пар підстав ділиться на 180-200. При электрофорезе фрагменти дають характерну картину сходів. Ця картина відрізняється від такої при некрозі кліток, де довжина фрагментів ДНК варіює. Фрагментація ДНК в нуклеосомах відбувається під дією кальцій чутливої эндонуклеазы. Ендонуклеаза в деяких клітках знаходиться постійно (наприклад, в тимоцитах), де вона активується появою в цитоплазмі вільного кальцію, а в інших клітках синтезується перед початком апоптоза. Проте ще не встановлено, яким чином після розщеплювання ДНК эндонуклеазой відбувається конденсація хроматина. Формування в цитоплазмі порожнин і апоптотических тілець. У апоптотической клітці спочатку формуються глибокі впячивания поверхні з утворенням порожнин, що приводить до фрагментації клітки і формування оточених мембраною апоптотических тілець, що складаються з цитоплазми і щільно розташованих органелл, з або без фрагментів ядра.
Фагоцитоз апоптотических кліток або тілець здійснюється навколишніми здоровими клітками, або паренхиматозными, або макрофагами. Апоптотічеськие тельця швидко руйнуються в лизосомах, а навколишні клітки або мігрують, або діляться, щоб заповнити простір, що звільнився після загибелі клітки.
Фагоцитоз апоптотических тілець макрофагами або іншими клітками активується рецепторами на цих клітках: вони захоплюють і поглинають апоптотические клітки. Один з таких рецепторів на макрофагах – рецептор витронектина, який є в3-интегрином і активує фагоцитоз апоптотических нейтрофілів.
Апоптоз бере участь в наступних фізіологічних і патологічних процесах:
запрограмованому руйнуванні кліток під час эмбриогенеза (включаючи імплантацію, органогенез). Не дивлячись на те, що при эмбриогенезе апоптоз не завжди є віддзеркаленням “запрограмованої смерті клітки”, це визначення апоптоза широко використовують різні дослідники
гормон-зависимой інволюції органів у дорослих, наприклад, відторгнення ендометрія під час менструального циклу, атрезії фолікулів в яєчниках в менопаузі і регресія молочної залози після припинення лактації
видаленні деяких кліток при проліферації клітинної популяції
загибелі окремих кліток в пухлинах, в основному при її регресії, але також і в пухлині, що активно росте
загибелі кліток імунної системи, як В-, так і Т-лимфоцитов, після виснаження запасів цитокинов, а також загибелі аутореактивных Т-клеток при розвитку в тимусе
патологічній атрофії гормон - залежних органів, наприклад, атрофії передміхурової залози після кастрації і виснаженні лімфоцитів в тимусе при терапії глюкокортикоїдами
патологічній атрофії паренхиматозных органів після обтурації вивідних проток, що спостерігається в підшлунковій і слинних залозах, нирках
загибелі кліток, викликаних дією цитотоксических Т-клеток, наприклад, при відторгненні трансплантата і хвороби “трансплантат проти господаря”
пошкодженні кліток при деяких вірусних захворюваннях, наприклад, при вірусному гепатиті, коли фрагменти апоптотических кліток виявляються в печінки, як тельця Каунсильмана
загибелі кліток при дії різних ушкоджувальних чинників, які здатні викликати некроз, але що діють в невеликих дозах, наприклад, при дії високої температури, іонізуючого випромінювання, протипухлинних препаратів.
Молекулярні механізми апоптоза. Апоптоз – багатоетапний процес. Перший етап – прийом сигналу, передвісника загибелі у вигляді інформації, що поступає до клітки ззовні або що виникає в надрах самої клітки. Сигнал сприймається рецептором і піддається аналізу.
Далі через рецептори або їх поєднання отриманий сигнал послідовно передається молекулам-посередникам (мессенджерам) різного порядку і зрештою досягає ядра, де і відбувається включення програми клітинного самогубства шляхом активації летальних і/або репресії антилетальних генів. Проте існування ПКС (програмована клітинна смерть) в без'ядерних системах (цитопластах – клітках, позбавлених ядра) показує, що наявність ядра не є обов'язковою для реалізації процесу.
Стосовно кліток тварин і людини апоптоз в більшості випадків пов'язаний з протеолітичною активацією каскаду каспаз – сімейства еволюційно консервативних цистеиновых протеаз, які специфічно розщеплюють білки після залишків аспарагиновой кислоти.
На основі структурної гомологии каспазы підрозділяються на підродини:
а) каспазы-1 (каспазы 1, 4, 5)
б) каспазы-2 (каспаза-2)
в) каспазы-3 (каспазы 3, 6–10) .
Цистєїновиє протеазы, мабуть, беруть участь також в ПКС у рослин. Проте апоптоз можливий і без участі каспаз: надсинтез белков-промоторов апоптоза Вах і Bak індукує ПКС у присутності інгібіторів каспаз.
В результаті дії каспаз відбувається:
активація прокаспаз з утворенням каспаз;
розщеплювання антиапоптозных білків сімейства Bcl-2. Піддається протеолизу інгібітор Днкази, відповідальний за фрагментацію ДНК. У нормальних клітках апоптозная Днказа CAD (caspase-activated DNase) утворює неактивний комплекс з інгібітором CAD, ICAD, що позначається. При апоптозе інгібітор ICAD за участю каспаз 3 або 7 инактивируется, і вільна CAD, викликаючи межнуклеосомальные розриви хроматина, веде до утворення фрагментів ДНК з молекулярною масою, кратній молекулярній масі ДНК в нуклеосомных частинках – 180-200 пар нуклеотидов. Апоптоз можливий і без фрагментації ДНК. Виявлений ядерний білок Acinus (apoptotic chromatin condensation inducer in the nucleus), з якого при комбінованій дії каспазы-3 і неідентифікованої протеазы утворюється фрагмент. Цей фрагмент у присутності додаткових неядерних чинників викликає апоптотическую конденсацію хроматина і фрагментацію ядра (кариорексис) без фрагментації ДНК;
гідроліз білків ламинов, що армують ядерну мембрану. Це веде до конденсації хроматина;
руйнування білків, що беруть участь в регуляції цитоскелета;
инактивация і порушення регуляції білків, що беруть участь в репарації ДНК, сплайсинге мРНК, реплікації ДНК.
Мішенню каспаз є поли(ADP-рибозо) полимераза (ПАРП). Цей фермент бере участь в репарації ДНК, каталізує поли(ADP-рибозилирование) білків, пов'язаних з ДНК. Донором ADP-рибозы є NAD+. Активність ПАРП зростає в 500 разів і більш при пов'язанні з ділянками розриву ДНК. Апоптотічеськая загибель клітки супроводжується розщеплюванням ПАРП каспазами. Надмірна активація ПАРП при масованих розривах ДНК, сильно знижуючи зміст внутріклітинного NAD+, веде до придушення гліколізу і мітохондріального дихання і викликає загибель клітки по варіанту некрозу.
Шляхи реалізації програми ПКС (програмована клітинна смерть).
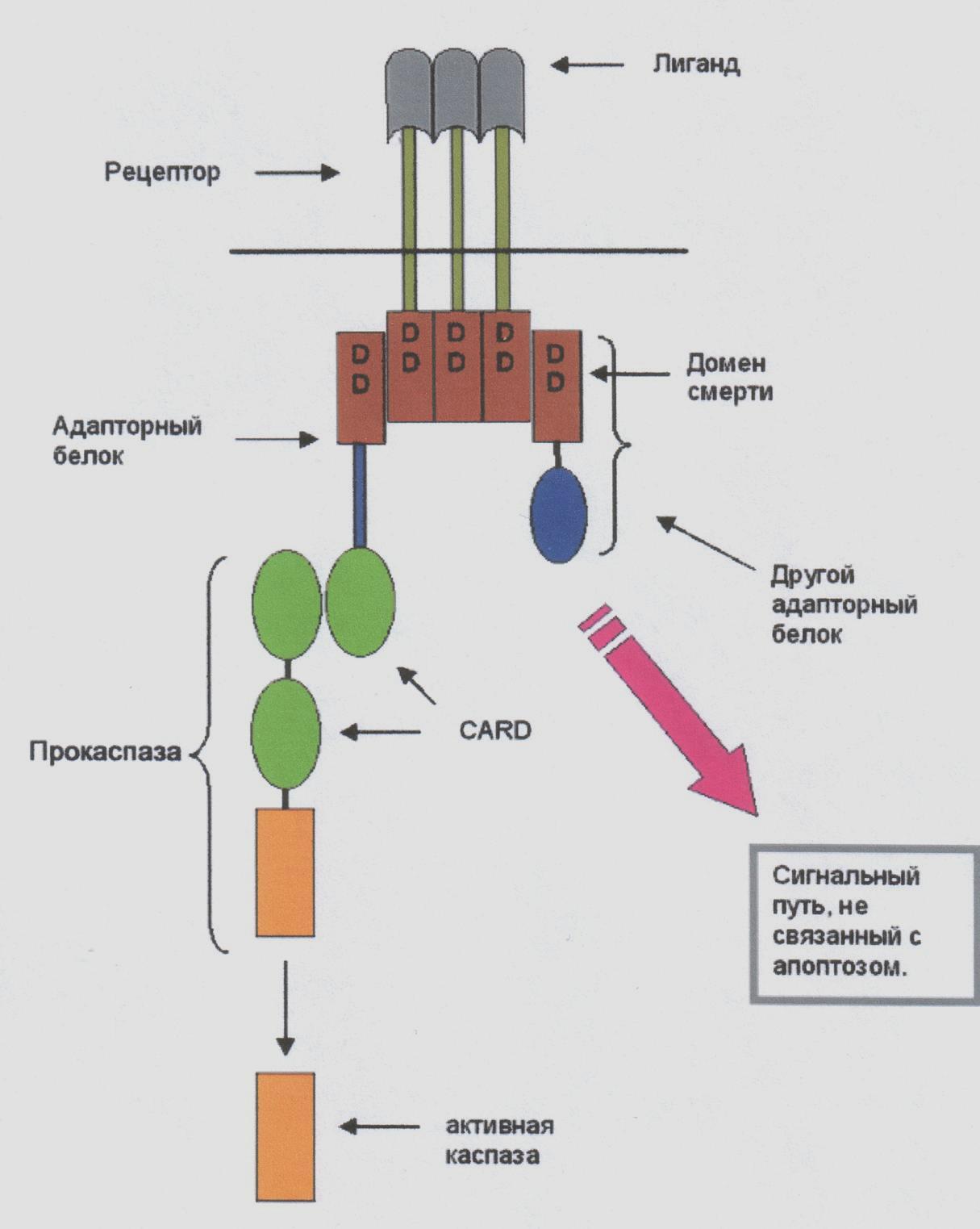
1. Серед них важливе місце займає шлях, опосередкований фізіологічними індукторами, дія яких реалізується через клітинні рецептори плазматичної мембрани, спеціально призначені для включення програми апоптоза. Цей шлях передачі сигналу ПКС схематично можна зобразити таким чином: індуктори ’ рецептори ’ адаптери ’ каспазы першого ешелону ’ регулятори ’ каспазы другого ешелону. Так, рецептор, Fas, що позначається, взаємодіючи з відповідним лигандом (лигандом FASL), трансмембранным білком Т-киллера, активується і запускає програму смерті клітки, інфікованої вірусом. Тим же шляхом при взаємодії з лигандом FASL на поверхні ТН-1-ЛИМФОЦИТОВ або з антитілом до Fas-рецептору гинуть ті, що стали непотрібними організму В-лімфоцити, що видужав, продуценти антитіл, що несуть Fas-рецептор. FASL– лиганд, що відноситься до численного сімейства чинника некрозу пухлин TNF. Це сімейство гомотримерных лигандов, окрім FASL і TNFa, включає TNFb (лимфотоксин).
Fas – член сімейства рецепторів TNF. Всі вони представлені трансмембранными білками, які позаклітинними ділянками взаємодіють з тримерами лигандов-індукторів. І їх взаємодія запускає в клетке- мішені процес апоптоза. Тому ж сприяє і білок перфорин, який виділяється Т-киллерами і утворює канали в мембрані клітки-мішені: через ці канали в клітку проникають протеолітичні ферменти гранзимы. Це так званий інструктивний апоптоз, який абсолютно необхідний для нормальної роботи імунної системи ссавців. Ключовими молекулами інструктивного апоптоза є рецептори смерті. Ліганди рецепторів смерті є триммеры. Тріммер лиганда зв'язується з трьома молекулами рецептора, викликаючи його триммеризацию. Рецептор має хвіст цитоплазми, що містить домени смерті. Від них сигнал поступає до адапторным білок, які активують каспазы, – ферменти апоптоза (рис.46.).
Одним з найбільш вивчених рецепторів смерті є рецептор Fas, що сприймає сигнал, який доходить каспазы, – 8. Він ініціює апоптоз, активуючи інші каспазы – эффекторные. Ці каспазы руйнують клітинні структури. Таким чином, каспаза – 8 + каспазы–эффекторные це «знаряддя» апоптоза, а ядерні і цитоплазма білки – «мішені» каспаз.
Fas і його лиганд (FAS-L) – важливі эффекторные молекули. Ними володіють і використовують їх цитотоксические иммуноциты в захисній реакції проти ракових кліток і кліток, уражених вірусами.
Щоб не допустити невчасний апоптоз, передачу сигналу через рецептори смерті контролюють ряд клітинних механізмів:
існують спеціальні білки, що перешкоджають спонтанній агрегації молекул рецептора;
існують рецептори-приманки, що не містять повноцінного домена смерті або позбавлені хвоста цитоплазми.
Fas і його лиганд також важливі для елімінації активованих иммуноцитов, що потрапили в иммуннопривилегированные органи, – семенники і очі.
Взаємодія рецептора і лиганда приводить до утворення кластерів рецепторних молекул і скріплення їх внутріклітинних ділянок з адаптерами.
Адаптер, зв'язавшись з рецептором, вступає у взаємодію з эффекторами, поки що неактивними попередниками протеаз з сімейства каспаз першого ешелону (каспаз, що ініціюють).
Взаємодія адаптера з рецептором і эффектором здійснюється через гомофильные белок-белковые взаємодії невеликих доменів: DD (death domain – домен смерті), DED (death-effector domain – домен эффектора смерті), CARD (– домен активації і рекрутування каспазы). Всі вони мають схожу структуру, містять по шість а-спиральных ділянок. Домени DD(домен смерті) беруть участь у взаємодії рецептора Fas з адаптером FADD (Fas-associated DD-protein). Домени DED беруть участь у взаємодії адаптера FADD з прокаспазами 8 і 10.
Ріс.46. Загальна схема передачі сигналу через рецептори смерті.
Найдетальніше охарактеризована прокаспаза-8, що рекрутує рецептором Fas через адаптер FADD. Утворюються агрегати FASL – Fas – FADD – прокаспаза-8. Подібні агрегати, в яких відбувається активація каспаз, названі апоптосомами, апоптозными шаперонами, або сигнальними комплексами, що індукують смерть.
Прокаспази володіють незначною протеолітичною активністю, що становить 1–2% активності зрілої каспазы. Будучи в мономірній формі, прокаспазы, концентрація яких в клітці нікчемна, знаходяться в латентному стані. Передбачається, що просторове зближення молекул прокаспаз при їх агрегації веде до утворення активних каспаз через механізм протеолітичного само- і перехресного розщеплювання (ауто- або транс-процессинга). В результаті від прокаспазы (молекулярна маса 30–50 кДа) відділяється регуляторний N-концевой домен (продомен), а частина молекули, що залишилася, розділяється на велику (~20 кДа) і малу (~10 кДа) субъединицы. Потім відбувається асоціація великою і малою субъединиц. Два гетеродимера утворюють тетрамер з двома каталітичними ділянками, що діють незалежно один від одного. Таким чином, прокаспаза-8 активується і вивільняється в цитоплазму у вигляді каспазы-8. Існують інші шляхи активації каспазы-8 – за участю рецепторів TNFR1 і DR3.
На етапі активації каспаз першого ешелону життя клітки ще можна зберегти. Існують регулятори, які блокують або, навпаки, підсилюють руйнівну дію каспаз першого ешелону. До них відносяться білки Bcl-2 (інгібітори апоптоза: A1, Bcl-2, BCL-W, BCL-XL, Brag-1, Mcl-1 і NR13) і Вах (промоторы апоптоза: Bad, Bak, Вах, BCL-XS, Bid, Bik, Bim, Hrk, Mtd). Ці білки еволюційно консервативні: гомолог Bcl-2 виявлений навіть у губок, у яких апоптоз необхідний для морфогенезу.
Каспаза-8 активує каспазу другого ешелону (эффекторную каспазу): шляхом протеолиза з прокаспазы-3 утворюється каспаза-3, після чого процес, запущений програмою смерті, виявляється необоротним.
Каспаза-3 здібна надалі до самостійної активації (автокаталізу або автопроцесингу), активує ряд інших протеаз сімейства каспаз, активує чинник фрагментації ДНК, веде до необоротного розпаду ДНК на нуклеосомальные фрагменти. Так запускається каскад протеолітичних ферментів, здійснюючих апоптоз.
2. Другий шлях реалізації програми ПКС пов'язаний з мітохондріальним цитохромом з. У клітках, що піддалися дії індуктора апоптоза, різко знижується мембранний потенціал (Dy) мітохондрій. Падіння Dy обумовлене збільшенням проникності внутрішньої мембрани мітохондрій унаслідок утворення гігантських пір. Існує багато чинників, що викликають розкриття пір. До них відносяться виснаження кліток відновленим глутатионом, NAD(P) H, ATP і ADP, утворення активних форм кисню, роз'єднування окислительного фосфорелирования протонофорными з'єднаннями, збільшення змісту Ca2+ в цитоплазмі. Утворення пір в мітохондріях можна викликати церамидом, NO, каспазами, амфипатическими пептидами, жирними кислотами. Пори мають діаметр 2,9 нм, що дозволяє перетинати мембрану речовинам з молекулярною масою 1,5 кДа і нижче. Наслідком розкриття пір є набухання мітохондріального матриксу, розрив зовнішньої мембрани мітохондрій і вивільнення розчинних білків міжмембранного об'єму. Серед цих білків – ряд апоптогенных чинників: цитохром з, прокаспазы 2, 3 і 9, білок AIF (apoptosis inducing factor), флавопротеин, що є, з молекулярною масою 57 кДа.
Утворення гігантських пір не є єдиним механізмом виходу міжмембранних білків мітохондрій в цитоплазму. Передбачається, що розрив зовнішньої мембрани мітохондрій може бути викликаний гіперполяризацією внутрішньої мембрани. Можливий і альтернативний механізм, без розриву мембрани, – розкриття гігантського білкового каналу в самій зовнішній мембрані, здатного пропускати цитохром і інші білки з міжмембранного простору.
Цитохром, що вивільняється з мітохондрій, з разом з чинником цитоплазми APAF-1 (apoptosis protease activating factor-1) бере участь в активації каспазы-9. APAF-1 – білок з молекулярною масою 130 кДа, що містить CARD-домен (caspase activation and recruitment domain) утворює комплекс з прокаспазой-9 у присутності цитохрома з і dATP або АТР. З цих субъединиц збираються жорсткі, симетричні структури, на зразок віяла або пропелера. APAF-1 грає роль арматури, на якій відбувається аутокаталитический процесинг каспазы-9. Передбачається, що в результаті залежного від гідролізу dATP (або АТР) конформаційної зміни APAF-1 набуває здатності пов'язувати цитохром (рис.47). Пов'язавши цитохром з, APAF-1 зазнає подальшу конформаційну зміну, сприяючу його олигомеризации і що відкриває доступ CARD-домена APAF-1 для прокаспазы-9, яка теж містить CARD-домен. Так утворюється конструкція, звана також апоптосомой, з молекулярною масою > 1,3 млн дальтон, у складі якої – не менше 8 субъединиц APAF-1. Завдяки гомофильному CARD-CARD-взаимодействию з APAF-1 в эквимолярном співвідношенні зв'язується прокаспаза-9, а потім прокаспаза-9 зв'язує прокаспазу-3. Просторове зближення молекул прокаспазы-9 на мультимерной арматурі з APAF-1-цитохром-с-комплексов, мабуть, приводить до міжмолекулярного протеолітичного процесингу прокаспазы-9 з утворенням активної каспазы-9. Зріла каспаза-9 потім розщеплює і активує прокаспазу-3.
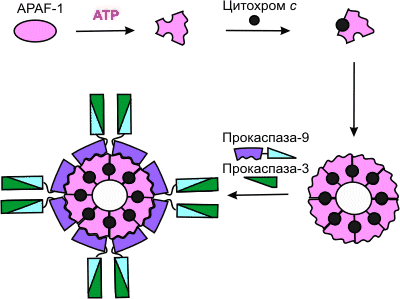
Ріс.47. Флавопротєїн - мітохондріальний эффектор апоптоза у тварин
Флавопротєїн AIF, будучи доданим, до ізольованих ядер з кліток HeLa, викликає конденсацію хроматина і фрагментацію ДНК, а при додаванні до ізольованих мітохондрій печінки щурів (вивільнення цитохрома з і каспазы) AIF є мітохондріальним эффектором ПКС, що діє незалежно від каспаз (рис.47).
Окрім розглянутих компонентів, при порушенні зовнішньої мембрани мітохондрій з міжмембранного об'єму виділяється термолабільний чинник, що викликає необоротне перетворення ксантиндегидрогеназы в ксантиноксидазу. Ксантіндегидрогеназа каталізує залежне від NAD+ окислення ксантина до гипоксантина і подальшого окислення гипоксантина до сечової кислоти. Ксантіноксидаза каталізує ті ж реакції, але не з NAD+, а з О2 як акцептор електронів. При цьому утворюються О2a, Н2о2, а з них – і інші активні форми кисню (АФК), які руйнують мітохондрії і є могутніми індукторами апоптоза. Механізми утворення АФК, звичайно, не обмежуються ксантиноксидазной реакцією. Головним джерелом АФК в клітках є мітохондрії. Різке збільшення АФК відбувається при зростанні мембранного потенціалу в мітохондріях, коли понижено споживання ATP і швидкість дихання лімітується ADP . Мембрана цитоплазми макрофагів і нейтрофілів містить О2a – генеруючу NADPH-оксидазу.
Залежно від шляху, по якому здійснюється активація каспаз, розрізняють різні типи кліток. Клітки типу I (зокрема, лінія лимфобластоидных В-клеток SKW і T-клетки лінії Н9) піддаються ПКС по шляху, залежному від апоптозных рецепторів плазматичної мембрани без участі мітохондріальних білків. Клітки типу II (наприклад, лінії Т-клеток Jurkat і СЕМ) гинуть по дорозі апоптоза, залежному від мітохондріального цитохрома з. ПКС, викликана з'єднаннями хіміотерапій, УФ- або і-облучением, мабуть, безпосередньо пов'язана з апоптозной функцією мітохондрій.
Деякі клітки, наприклад, клітки ембріональної нервової системи, включають механізми апоптоза, якщо вони відчувають дефіцит апоптозподавляющих сигналів (званих також чинниками виживання) від інших кліток. Фізіологічний сенс процесу – в елімінації надмірних нервових клітин, що конкурують за обмежений фонд чинників виживання. Епітеліальні клітки при відділенні від позаклітинного матриксу, що виробляє чинники виживання, теж приречені на ПКС. Чинники виживання зв'язуються відповідними рецепторами цитоплазми, активуючи синтез переважних апоптоз агентів і блокуючи стимулятори апоптоза. Деякі речовини (наприклад, стероїдні гормони) надають диференційований ефект на різні типи кліток – запобігають апоптоз одних типів кліток і індукують його у інших. Так, за наявності в позаклітинному матриксі чинників зростання PDGF (platelet-derived growth factor – тромбоцитарный чинник зростання) або NGF (nerve growth factor – чинник зростання нервів) і цитокина інтерлейкіна-3 (IL-3) проапоптозный білок Bad не активний.Чинники зростання, зв'язавшись зі своїм рецептором на плазматичній мембрані, викликають активацію цитозольной протеинкиназы В, і що каталізує фосфорилування Bad по Ser-136. IL-3 теж зв'язується зі своїм рецептором на плазматичній мембрані і активує мітохондріальну cAMP-зависимую протеинкиназу А, що каталізує фосфорилування Bad по Ser-112. Будучи фосфорилированным по обох залишках серина, Bad утворює комплекс з білком 14-3-3, розташований в цитоплазмі. Дефіцит чинників зростання і IL-3 сприймається кліткою як сигнал до апоптозу: відбувається дефосфорилирование Bad, його впровадження в зовнішню мембрану мітохондрій, вихід цитохрома із з мітохондрій і подальша активація каспазы-9 через APAF-1-зависимый механізм.
У ряді випадків ПКС реалізується в результаті комбінованої дії двох шляхів – з участю і рецепторів плазматичної мембрани, і мітохондріального цитохрома з. Так, пошкодження ДНК веде до накопичення в клітці білкового продукту гена р53, який може зупиняти ділення клітин і/або індукувати апоптоз. Білок р53 є чинником транскрипції, регулюючим активність ряду генів. Передбачається, що у відповідь реакція на утворення білка р53 залежить від ступеня порушення клітинного генома. При помірному порушенні генома відбувається зупинка клітинного ділення, здійснюється репарація ДНК, і клітка продовжує своє існування. При надмірному порушенні генома, коли ДНК вже не піддається репарації, включаються рецепторний і цитохром с-зависимый апоптозные каскади активації каспаз.
Також існує шлях передачі сигналу ПКС за участю эндоплазматического ретикулума (ЕР). У ЕР локалізована прокаспаза-12. Порушення внутріклітинного Ca2+-гомеостаза добавкою тапсигаргина або Ca2+ -ионофорного антибіотика А23187 веде до апоптозу кліток, викликаному перетворенням прокаспазы-12 в каспазу-12. ЕР-ЗАВІСИМИЙ апоптоз пов'язаний з хворобою Альцгеймера.
Цитотоксичеськие лімфоцити, Т-киллеры, можуть викликати апоптоз у інфікованих кліток за допомогою білка перфорина. Полімеризуючись, перфорин утворює в мембрані цитоплазми клітки-мішені трансмембранные канали, по яких всередину клітки поступають TNFb, гранзимы (фрагментины) – суміш сериновых протеаз. Істотним компонентом цієї суміші є гранзим В – протеолітичний фермент, перетворюючий прокаспазу-3 в активну каспазу-3 .
Взаємодія кліток з позаклітинним матриксом здійснюється за допомогою интегринов. Інтегріни – велике сімейство гетеродимерных мембранних білків, які беруть участь в адгезії кліток, пов'язуючи внутріклітинний цитоскелет з лигандами позаклітинного матриксу. Порушення адгезії кліток індукує апоптоз.
Особливу форму апоптоза зазнають еритроцити ссавців. Біогенез еритроцитів з плюрипотентной стволової клітини в кістковому мозку включає ряд проміжних етапів. На етапі эритробласта ядро виганяє (виштовхується) з клітки і пожирається макрофагом. Альтернативний варіант: кариорексис (деструкція ядра) з освітою телець Жоллі і їх подальший розпад і лізис усередині клітки. Без'ядерна клітка, звана ретикулоцитом, надалі втрачає мітохондрії і рибосомы і перетворюється на еритроцит. Втрату ядра эритробластом можна розглядати як особливу форму ядерного апоптоза. З'ясування його механізму дозволило б застосувати його для знешкодження пухлинних кліток.
Регуляція апоптоза. Апоптоз це генетично контрольована смерть клітки. В даний час виявлено велике число генів, які кодують речовини, необхідні для регуляції апоптоза. Багато хто з цих генів зберігся в ході еволюції - від круглих черв'яків до комах і ссавців. Деякі з них виявляються також в геномі вірусів. Таким чином, основні біохімічні процеси апоптоза в різних експериментальних системах (дослідження ведуться на круглих черв'яках і мухах) є ідентичними, тому результати досліджень можна прямо переносити на інші системи (наприклад, організм людини).
Апоптоз може регулюватися:
зовнішніми чинниками
автономними механізмами.
Дія зовнішніх чинників. Апоптоз може регулюватися дією багатьох зовнішніх чинників, які ведуть до пошкодження ДНК. При невідновленому пошкодженні ДНК шляхом апоптоза відбувається елімінація потенційно небезпечних для організму кліток. У даному процесі велику роль грає ген супрессии пухлин р53. До активації апоптоза також приводять вірусні інфекції, порушення регуляції клітинного зростання, пошкодження клітки і втрата контакту з тими, що оточують або основною речовиною тканини. Апоптоз – це захист організму від персистенції пошкоджених кліток, які можуть опинитися потенційно небезпечними для багатоклітинного організму. При стимуляції тканин яким-небудь митогеном її клітки переходять в стан підвищеної митотической активності, яка обов'язково супроводжується деякою активацією апоптоза. Доля дочірніх кліток (виживуть вони або піддадуться апоптозу) залежить від співвідношення активаторів і інгібіторів апоптоза: - інгібітори включають чинники зростання, клітинний матрикс, статеві стероїди, деякі вірусні білки; - активатори включають недолік чинників зростання, втрату зв'язку з матриксом, глюкокортикоїди, деякі віруси, вільних радикалів, іонізуючу радіацію.
При дії активаторів або відсутності інгібіторів відбувається активація ендогенних протеаз і эндонуклеаз. Це приводить до руйнування цитоскелета, фрагментації ДНК і порушення функціонування мітохондрій. Клітка зморщується, але клітинна мембрана залишається интактной, проте пошкодження її приводить до активації фагоцитозу. Загиблі клітки розпадаються на невеликі, оточені мембраною, фрагменти, які позначаються як апоптотические тільця. Запальна реакція на апоптотические клітки не виникає.
Автономний механізм апоптоза. При розвитку ембріона розрізняють три категорії автономного апоптоза: морфогенетический, гистогенетический і філогенез.
Морфогенетічеський апоптоз бере участь в руйнуванні різних тканинних зачатків. Прикладами є:
руйнування кліток в межпальцевых проміжках;
загибель кліток приводить до руйнування надмірного епітелію при злитті піднебінних відростків, коли формується тверде небо.
загибель кліток в дорсальній частині нервової трубки під час зімкнення, що необхідне для досягнення єдності епітелію двох сторін нервової трубки і пов'язаної з ними мезодермы.
Гистогенетічеський апоптоз спостерігається при диференціюванні тканин і органів, що спостерігається, наприклад, при гормональнозависимой диференціюванні статевих органів з тканинних зачатків. Так, у чоловіків клітками Сертолі в яєчках плоду синтезується гормон, який викликає регресію проток Мюллера (з яких у жінок формуються маткові труби, матка і верхня частина піхви) шляхом апоптоза.
Філогенез апоптоз бере участь у видаленні рудиментарних структур у ембріона, наприклад, пронефроса.
При різних станах може спостерігатися як прискорення, так і уповільнення апоптоза. Не дивлячись на те, що апоптоз можуть активувати різні чинники, характерні для певних типів кліток, проте кінцевий шлях апоптоза регулюється точно встановленими генами і є загальним, незалежно від причини активації апоптоза. Всі чинники, що підсилюють або ослабляючі апоптоз, можуть діяти прямо на механізм загибелі клітки або опосередковано, шляхом впливу на регуляцію транскрипції. В деяких випадках вплив цих чинників на апоптоз є вирішальним (наприклад, при глюкокортикоид- залежному апоптозе тимоцитов), а в інших не має особливої важливості (наприклад, при Fas- і TNF-зависимом апоптозе). В процесі регуляції бере участь велика кількість речовин. Найбільш вивченими з них є речовини з сімейства bcl-2. Bcl-2 ген вперше був описаний як ген, який транслоцируется в клітках фолікулярної лімфоми і інгібірує апоптоз. При подальших дослідженнях виявилось, що Bcl-2 є мультигеном, який виявляється навіть у круглих черв'яків. Гомологичниє гени були також виявлені в деяких вірусах. Всі речовини, що відносяться до даного класу, діляться на активатори і інгібітори апоптоза.
До інгібіторів відносяться: bcl-2, bcl-xL, Mcl-1, bcl-w, аденовирусный E1B 19K, Эпштейн-Барр-вирусный BHRF1. До активаторів відносяться bax, bak, Nbk/Bik1, Bad, bcl-xS. Члени цього сімейства взаємодіють один з одним. Одним з рівнів регуляції апоптоза є взаємодія білок-білок. Білки сімейства bcl-2 формують як гомо- так і гетеродимеры. Наприклад, bcl-2-ингибиторы можуть утворити димеры bcl-2-активаторами. Таким чином, життєздатність кліток залежить від співвідношення активаторів і інгібіторів апоптоза. Наприклад, bcl-2 взаємодіє з bax; при переважанні першого життєздатність клітки підвищується, при надлишку другого – зменшується. До того ж білки сімейства bcl-2 можуть взаємодіяти з білками, що не відносяться до цієї системи. Наприклад, bcl-2 може з'єднаються з R-ras, який активує апоптоз. Інший білок, Bag-1, підсилює здатність bcl-2 інгібірувати апоптоз.
В даний час прийнято вважати, що гени, що беруть участь в регуляції зростання і розвитку пухлин (онкогены і гены-супрессоры пухлин), грають регулюючу роль в індукції апоптоза. До них відносяться:
bcl-2 онкоген, який інгібірує апоптоз, викликаний гормонами і цитокинами, що приводить до підвищення життєздатності клітки;
білок bax (також з сімейства bcl-2) формує димеры bax-bax, які підсилюють дію активаторів апоптоза. Відношення bcl-2 і bax визначає чутливість кліток до апоптотическим чинників і є “молекулярним перемикачем”, який визначає, чи відбуватиметься зростання або атрофія тканини;
c-myc онкоген, чий білковий продукт може стимулювати або апоптоз, або зростання кліток (за наявності інших сигналів виживання, наприклад, bcl-2);
ген р53, який в нормі активує апоптоз, але при мутації або відсутності (що виявлене в деяких пухлинах) підвищує ту, що виживає кліток. Встановлено, що р53 необхідний для апоптоза при пошкодженні клітки іонізуючим випромінюванням, проте при апоптозе, викликаному глюкокортикоїдами і при старінні, він не потрібний.
Зниження апоптоза. Продукт р53 гена стежить за цілісністю генома при митозе. При порушенні цілісності генома клітка перемикається на апоптоз. Навпаки, білок bcl-2 інгібірує апоптоз. Таким чином, недолік р53 або надлишок bcl-2 приводить до накопичення кліток: ці порушення спостерігаються в різних пухлинах.
Ослаблення апоптоза викликає патологічні процеси. Наприклад, приводить до утворення злоякісної пухлини. Спочатку мутації 5-8 экзонов гена р53, який зазвичай трансформує з нерепарированных розривах ланцюгів ДНК в сигнал до розвитку апоптоза, що приводить до елімінації кліток з втратою ДНК. У нормальних клітках білок р53 не виявляється, а при пухлинах його форму мутанта экспрессируют до 70% кліток. Без контролю гена р53 пухлинні клітки прогресують.
Зміни гена р53, гиперэкспресия ще двох генів (Вс1-1, Вс1-х), активація каспаз є основою формування стійкості до лікувальних дій, яка заснована на індукції апоптоза пухлинних кліток, – рентгено- і радіотерапії, хіміопрепаратам.
У результаті – виникає множинна лікарська стійкість пухлинних кліток.
Вивчення чинників регулюючих апоптоз має велике значення в розробці лікарських препаратів, що підсилюють загибель кліток злоякісних новоутворень. Аутоіммунні захворювання можуть відображати порушення в індукції апоптоза лімфоїдних кліток, здатних реагувати з власними антигенами. Наприклад, при системному червоному вовчаку спостерігається порушення Fas-рецепторов на клітинній поверхні лімфоцитів, що веде до активації апоптоза. Деякі віруси підвищують ту, що свою виживає шляхом ингибирования апоптоза інфікованих кліток, наприклад, вірус Епштейна-барра може впливати на обмін bcl-2.
Посилення апоптоза. Прискорення апоптоза доведене при синдромі придбаного імунодефіциту (СНІД), нейротрофических захворюваннях і деяких захворюваннях крові, при яких спостерігається дефіцит яких-небудь формених елементів. При СНІДІ вірус імунодефіциту може активувати CD4 рецептор на неінфікованих Т-лимфоцитах, прискорюючи, таким чином, апоптоз, що приводить до виснаження кліток даного типу.
Значення апоптоза в розвитку організму і патологічних процесах. Апоптоз грає важливу роль в розвитку ссавців і в різних патологічних процесах. Функціонування bcl-2 потрібне для підтримки життєздатності лімфоцитів, меланоцитов, епітелію кишечника і кліток нирок під час розвитку ембріона. bcl-x необхідний для ингибирования смерті кліток в эмбриогенезе, особливо в нервовій системі. Вах необхідний для апоптоза тимоцитов і підтримки життєздатності сперматозоїдів під час їх розвитку. р53 є геном супрессии пухлин, тому в эмбриогенезе особливої ролі не грає, але обов'язково необхідний для супрессии пухлинного зростання. Миші, у яких відсутні обидва р53 гена, проявляли надзвичайно високу схильність до розвитку злоякісних пухлин в результаті повного або часткового порушення апоптоза передпухлинних кліток. Посилений синтез білка, що кодується bcl-2 геном, приводить до придушення апоптоза і, відповідно, розвитку пухлин; даний феномен виявлений в клітках В-клеточной фолікулярної лімфоми. При лимфопролиферативных захворюваннях і схожій на системний червоний вовчак хворобі у мишей спостерігається порушення функції Fas-лиганда або Fas-рецептора. Підвищений синтез Fas-лиганда може попереджати відторгнення трансплантата. Апоптоз є частиною патологічного процесу при інфікуванні клітки аденовірусами, бакуловирусами, ВІЧ і вірусами грипу. Інгибірованіє апоптоза спостерігається при персистировании інфекції, в латентному періоді, а при посиленій реплікації аденовірусів, бакуловирусов, можливо герпесвирусов, вірусу Епштейн-Барра і ВІЧ спостерігається активація апоптоза, що сприяє широкому розповсюдженню вірусу. При нейродистрофічних захворюваннях наголошується порушення функції гена (iap-гена), схожого з інгібітором апоптоза бакуловирусов.
Апоптоз, некроз і проліферація кліток. Завдяки проточній цитометрии дослідники можуть легко відрізняти живі нейрони від тих, які встали на шлях клітинної смерті, і диференціювати некротичні нейрони від апоптозных на найраніших стадіях. Апоптоз - генетично запрограмована смерть, здійснювана за допомогою специфічних механізмів і ферментів. При апоптозе клітка зморщується, її структури руйнуються цистеиновыми-аспарагиновыми протеиназами, так званими каспазами. Сімейство цих ферментів (у нього входить близько десяти різних протеиназ) складає каскад взаимоконтролируемых білків, переклад яких в активний стан вимагає одночасної присутності ряду клітинних чинників. Такий ступінчастий механізм оберігає від випадкового виникнення апоптоза.
Некроз обумовлений механічним або іншим пошкодженням клітинної мембрани, порушенням цілісності і керованості клітки. Клітки, не здатні виконувати свої функції, вмирають, а їх велика кількість створює в тканині вогнище запалення.
Не дивлячись на принципові відмінності апоптоза і некрозу, їх об'єднує корисна властивість - вони допомагають організму очиститися від непотрібних (пошкоджених) або шкідливих (чужорідних) структур. У вогнище запалення спрямовуються макрофаги і інші клітки, “сміттярі”, що видаляють некротичні частини тканин або чужорідні частинки (наприклад, занози, що потрапили в тканині). За допомогою апоптоза організм намагається розпізнати і ліквідовувати клітки-мутанти, що стали небезпечними для організму (що перероджуються спонтанно або під впливом зовнішніх чинників). Так, частота появи в організмі злоякісних кліток багато вище, ніж вірогідність самого захворювання, оскільки в більшості випадків вони розпізнаються і нейтралізуються імунною системою без шкоди для організму.
Апоптоз запрограмований на поступове контрольоване усунення кліток, а некроз здійснюється швидко, хаотично і некеровано. При апоптозе фрагменти кліток або навіть цілі білкові молекули можуть використовуватися іншими клітками для виконання тих же самих функцій. Наприклад, в тимусе, де відбувається дозрівання лімфоцитів, клітки, що розпадаються при апоптозе, поставляють свої білки-рецептори для перетворення “юних” лімфоцитів на повноцінні імунні клітки.
Епітеліальні клітки слизової оболонки запрограмовані таким чином, що апоптоз індукується в них періодично і з великою частотою (вони живуть лише 1,5-2 тижні). Відторгнення апоптозных кліток знижує вірогідність проникнення в організм вірусної інфекції. Цікаво, що в російській армії для запобігання кишковим епідеміям по указу Петра I в їжу додавали перець. Сьогодні відомо, що це прекрасний засіб для активації апоптоза кліток слизистого епітелію.
Так або інакше, вигода розпізнавання ранніх стадій і типу клітинної смерті очевидна. Для кожного з них є свої специфічні маркери. Один з фосфоліпідів клітинних мембран, фосфатидилсерин, в нормальних умовах розташований з внутрішньої сторони мембранного бислоя, при порушеннях цитоскелета сигналізує про початок апоптоза. До речі, саме так макрофаги розпізнають і видаляють злоякісні клітки. Білки, чутливі до фосфатидилсерину (аннексины), використовують для раннього розпізнавання апоптозных кліток. А для некротичних кліток з пошкодженою мембраною є інший маркер. Їм може бути фарбник, наприклад иодид пропидия (PI), який зв'язується з нуклеїновими кислотами, але не проникає через мембрану живих (нативных) клітин.
Експериментально показано, що після тривалої (30 мін) індукції окислювального стресу активацією NMDA-рецепторов з'являються і некротичні, і апоптозные клітки, причому їх частку в популяції легко розрахувати. Таким чином, в руках дослідників є модель, що дозволяє оцінювати як потенційну уразливість нейронів з боку різних чинників, так і можливість захисту кліток від апоптоза або некрозу (наприклад, за допомогою лікарських препаратів).
Стежити за розвитком апоптоза можна також, вимірюючи активність внутріклітинних каспаз, які в клітці взаємно контролюють один одного. Так, при пов'язанні на клітинній мембрані позаклітинних сигнальних молекул із спеціальним рецептором (CD95/Fas) в цитоплазмі неактивна прокаспаза 8 перетворюється на активний фермент, який, у свою чергу, активує каспазу 3, що відкриває клітці шлях до апоптозу. Навантажуючи клітки флуорогенным субстратом каспазы 3 і стимулюючи їх різними способами, можна вимірювати сигнал від флуоресцентного продукту. Росте продукт - активується каспаза 3, і інтенсивність сигналу буде пропорційна активації ферменту і вірогідності розвитку апоптоза.
Проте каспаза 3 бере участь не тільки в реалізації апоптоза, але і в багатьох стадіях клітинного циклу і в процесах проліферації. Особливо важливі ці реакції для кліток імунної системи. Значить, у ряді випадків активність каспазы 3 не обов'язково означає початок апоптоза, а може бути пов'язана з проліферацією лімфоцитів.
Старіння і апоптоз. Відомий американський учений Л.Хейфлік в Медичному центрі дитячої лікарні Північної Кароліни вперше довів, що природна тривалість життя людини обумовлена числом митозов, яке можуть зробити клітки даного організму. Він брав шматочки шкіри від ембріона, новонародженої і дорослої людини, розбивав їх на окремі клітки і культивував в спеціальному живильному середовищі. Виявилось, що клітки ембріона можуть зробити близько 50 ділень, а потім в них спостерігаються всі ознаки апоптотической смерті. У дорослої людини клітки могли зробити вже не 50, а значно менше ділень, залежно від віку обстежуваного пацієнта. Згодом було показано, що механізм старечого апоптоза запускається і знаходитися в ядрі.
В даний час для пояснення молекулярно-генетичних механізмів старіння організму запропоновано три гіпотези.
1. Перша гіпотеза особливо виразно розвинена в працях професора Ж. Медведева, а також Л. Орджелом з Інституту ним. Соління в США. Ці дослідники вважають, що старіння це процес накопичення помилок в процесах транскрипції і трансляції і виникненні ферментів з дефектним функціонуванням. При цьому механізми репарації не можуть справиться зі все зростаючою кількістю дефектів.
2. Згідно другій гіпотезі, запропонованій також Ж.Медведевим 0,4% інформації що міститься в ДНК клітинного ядра, використовується кліткою постійно впродовж її життя. Крім того, багато генів в молекулі ДНК повторюються, роблячи генетичну інформацію у високому ступені надмірної. Ж. Медведев припустив, що послідовності, що повторюються, зазвичай репресовані, але у разі значного пошкодження активного гена він замінюється одним з ідентичних резервних генів. Надмірність ДНК може, отже, служити гарантією проти внутрішньо властивої схильності системи випадковим молекулярним пошкодженням. Проте поступово весь резерв генів буде вичерпаний і тоді починають виникати патофізіологічні зміни, які приведуть до загибелі клітки. Таким чином, ніж більше надмірної ДНК, тим більше тривалість життя даного вигляду.
3. Третя гіпотеза постулювала, що вікові зміни є продовженням нормальних генетичних сигналів, регулюючих розвиток тварини від моменту його зачаття до статевого дозрівання. Мабуть, є «гени старіння», які уповільнюють або навіть закривають біохімічні шляхи один за іншим і ведуть до передбачених вікових змін. При цьому знижуються функціональні можливості кліток. Старіння організму - це по суті старіння і апоптоз ключових кліток, загибель яких здатна вплинути на фізіологію всього організму.
Фармакологічна корекція апоптоза. Численні дослідження останніх років показали, що часто саме апоптоз, а не некроз лежить в основі інфаркту міокарду, гострої ниркової недостатності, інсульту, травми головного і спинного мозку і інших захворювань, пов'язаних з високою смертністю.
Відомо декілька генів, відповідальних за розвиток апоптоза в спинному мозку: індуктори – Fas/apo-1, p53; інгібітори - Bcl-2, bax. Добре вивчені гени ранньої негайної відповіді і відповідні ним білки. Зокрема, це білок р53, відомий, як регулятор клітинного циклу і супрессор пухлин, який бере участь у відновленні ДНК - пошкодженої клітки.
Як відомо, апоптоз має дві стадії: оборотну (початкова) і необоротну. Розробка терапевтичної дії на апоптоз знаходиться на початковому етапі. Знання тимчасової характеристики розвитку апоптоза дозволяє прогнозувати і планувати лікувальну практику. Встановлено, що метилпреднизолон пригнічує апоптоз на віддаленні вогнища травми, рилузол – блокує протеолиз цитоскелета.
Нову стратегію в розробці методів лікування пошкодженого спинного мозку представляє придушення експресії проапоптозных генів (р53, bcl-x) за допомогою антисмислових олигодезоксинуклеотидов. Вони інгібірують трансляцію мРНК.
Проблемними є: нестійка олигодезоксинуклеотидов; їх доставка в клітку.
Інший підхід до антиапоптозному лікування травми спинного мозку – генна терапія.
У зв'язку з апоптозом виникла необхідність перегляду ряду концептуальних основ фізіології і патології. Замість колишніх уявлень про загибель багатоклітинного організму, як негативному після значущості і часто випадкового явища, що ідентифікується з некрозом, сформувався новий погляд, згідно якому загибель частини кліток в межах організму є закономірним і необхідним явищем, і само існування багатоклітинного організму має на увазі баланс життя і смерті на рівні складових його клітинних популяцій.
Генетичні механізми канцерогенезу (нерегульоване клітинне зростання: клінічне значення).
Отже, ми визначили молекули, провідні клітку по фазах клітинного циклу, включаючи G1, G1/S, S і S/G2. Це складно взаємодіючі молекули, які балансують між фосфорилированным і дефосфорилированным станом, і таким чином сигналізують про початок процесу. Звичайно, для нормального перебігу клітинного циклу украй важлива присутність субъединиц комплексів (Cdk-циклин) і різних ингибиторных білків.
У нормі протягом эмбриогенеза і розвитку відбувається швидка проліферація кліток. Ми знаємо, що клітки можуть втрачати свою здатність контролювати клітинний цикл, що приводить до нерегульованого клітинного зростання, тобто до раку.
У всіх тканинах постійно йдуть процеси фізіологічної регенерації, яка супроводжується діленням і збільшенням кількості кліток. Порушення цих процесів приводить до утворення пухлини. Вони бувають доброякісні і злоякісні. Тип пухлини, що розвивається, багато в чому залежить від природи уражених генів.
Клітки злоякісної пухлини втрачають диференціювання, швидко розмножуються і розповсюджуються по організму, утворюючи вторинні пухлини. Крім того, ці клітки виділяють продукти розпаду.
Доброякісні пухлини не втрачають диференціювання, і не розповсюджуються по організму (фіброма – пухлина сполучної тканини, міома – пухлина м'язової тканини, папілома – пухлина шкіри). Проте ці пухлини можуть ставати злоякісними.
Під дією канцерогенів (випромінювання, хімічні речовини, онковирусы і ін.) клітка стає злоякісною. Пухлинні клітки мають змінений геном, в якому мутації зачіпають не одиничний ген, а цілу сукупність генів.
Тому процес пухлинного переродження – не одномоментна мутація, а тривале накопичення генетичних дефектів. І лише коли їх сума перевищує критичну межу, клітка перетворюється на пухлинну.
Всього в наших клітках — приблизно по 25 000 генів. До онкогенезу ж, у всіх його можливих проявах, мають відношення, за нинішніми даними, не більше 120-150 генів людини. Плюс деяка кількість вірусних генів. Всі гени, які можуть відповідати за онкогенез, ділять на декілька типів.
По-перше, це так звані мутаторные гени. При зниженні їх активності різко зростає швидкість накопичення мутацій і інших змін генома. Очевидно, до даного типу відносяться гени систем контролю за станом ДНК і репарації її пошкоджень. При порушенні їх дії розвивається пігментна ксеродерма і пухлини шкіри.
По-друге, це деякі гени вірусного походження, тобто вірусні онкогены.
Вже чітко доведено, що цілий ряд вірусів може викликати (хоча, звичайно, далеко не завжди викликає) пухлинне переродження зараженої клітки.
Причому є одна загальна для всіх цих вірусів особливість: пухлинному переродженню клітки передує вбудовування (інтеграція) вірусних генів (в т.ч. і онкогенов) в одну з її хромосом.
Проте інтеграція вірусних онкогенов в клітинну хромосому зовсім не означає, що клітка негайно перетвориться на пухлинну. Нерідко спочатку проходить досить тривалий (до декількох років) латентний період. Більш того, часом переродженню піддається навіть не та клітка, яка була інфікована, а одна з кліток наступних поколінь.
Все це і говорить про те, що одного лише впровадження вірусних онкогенов в хромосоми господаря недостатньо для трансформації клітки. Потрібна ще серія якихось (мабуть, випадкових, але достатньо визначених) змін клітинного генома. Відомо більше 50 ретровірусів, що викликають онкогенез.
По-третє, це протоонкогены. Так називаються цілком нормальні гени клітки (мало того, необхідні їй для забезпечення ряду ключових процесів), які, проте, стають украй небезпечними при неконтрольованій експресії або модифікації функцій.
Таким чином, зміна структури самі цих генів або якихось інших генів, контролюючих їх активність, перетворює протоонкогены в сьогодення клітинні онкогены. Іноді цьому перетворенню можуть сприяти і вірусні онкогены. До протоонкогенам відноситься ген циклина Д. Его гіперекспресія (ампліфікація) викликає рак молочних і слинних залоз.
По-четверте, це т.з. пухлинні супрессоры. Тут ситуація зворотна: потенційно небезпечно (у плані онкогенезу) не безконтрольне посилення функції такого гена, а навпаки — виключення функції. Що знову-таки відбувається унаслідок тієї або іншої генетичної перебудови або мутації. До пухлинних супрессорам відноситься ген р53.
В кінці 60-х років XX століття при гібридизації злоякісних кліток з нормальними була виявлена супрессия (придушення) злоякісних властивостей клітки. Повернення клітки до необмеженого розмноження відбувалося після втрати певної хромосоми. Це дозволило сформулювати поняття про наявність в геномі особливої групи генів, названих антионкогенами, або генами-супрессорами пухлин. Це аутосомно-домінантні гени.
Функція антионкогенов — пригнічувати проліферацію кліток, якщо вона виникне в невідповідному місці і в невідповідний час. Якщо в обох локусах (материнського і батьківського походження) антионкоген буде мутантом або делетированным, то починається неконтрольована проліферація кліток. Спочатку цитогенетичними, а пізніше молекулярно-генетичними дослідженнями встановлені наступні закономірності дії генов-супрессоров пухлин.
Індивід успадковує від одного з батьків мутацію в локусе гена-супрессора. Це може бути один з варіантів генної мутації або микроделеция. Гетерозіготность індивіда по даному локусу страхує його від виникнення пухлини, оскільки ген-супрессор — домінантний ген. Проте в процесі життя в соматичних клітках йде мутаційний процес, зокрема можуть виникнути мутації в «здоровій» хромосомі. Клітка буде гомозиготна, і, отже, знімається супрессия онкогенності. Це явище називається втратою конституціональної (природженою) гетерозиготности. У людини вже описано багато синдромів, при яких втрата гетерозиготности веде до злоякісних новоутворень. Для багатьох з цих форм відома не тільки хромосомна локалізація гена, але і його структура, мутації і первинні продукти дії генів. Проте для виникнення однієї і тієї ж пухлини необхідна втрата гетерозиготности не в одному, а в декількох локусах. Крім того, потрібні ще мутації в онкогенах. Многокомпонентность генетичних подій очевидна.
Відмітимо: мутаторные гени (тобто гени систем контролю за станом ДНК і репарації її пошкоджень) формально підпадають під визначення пухлинних супрессоров. При їх виключенні ризик трансформації клітки різко зростає.
І все-таки доцільно виділяти мутаторные гени серед пухлинних супрессоров через їх персоною функціональної ролі. Хоча, повторюваний, це виділення часом є достатньо умовним.
Всього відомо близько 100 протоонкогенов і порядка 20 пухлинних супрессоров. Це гени білків, що беруть (як «приводні паси» або стримуючі чинники) участь в таких процесах, як репарація ДНК, клітинний цикл, апоптоз і диференціювання кліток. Через те всі ці процеси зав'язані з онкогенезом в єдиний вузол (мал. 48).


Ріс.48. Порушення регуляції клітинного циклу і розвиток раки
Канцерогенні чинники. У 2000 році в світі, за даними Міжнародного агентства по дослідженню раки (МАЇР) (Ліон), було зареєстровано більше 10 млн. випадків захворювання злоякісними пухлинами (ЗО), а в 2020 році число знов виявлених випадків ЗО досягне 16 млн. Зростання кількості хворих ЗО в світі відбувається як в результаті збільшення чисельності населення, так і його старіння. Причиною збільшення числа хворих ЗО є також розповсюдження в світі і, особливо в країнах чинників навколишнього середовища і образу, що розвиваються, життя, які є причиною раки. В той же час в більшості розвинених країн знижуються стандартизованная за віком захворюваність і смертність на 100 тис. населення від багатьох форм ЗО. Зниження захворюваності ЗО, як відомо, досягається в першу чергу первинною профілактикою. Зниження ж смертності відбувається в результаті зменшення захворюваності, а також поліпшення тієї, що виживає, що у свою чергу може бути результатом удосконалення методів діагностики і лікування.
Прийнято вважати, що приблизно в 90% випадків виникнення раки обумовлене дією канцерогенних речовин в навколишньому середовищі. Канцерогени виявляють не тільки поблизу місць їх викидів, але і далеко за їх межами. Повсюдна їх присутність показує, що ізолювати людину від них практично неможливо: забруднення середовища носить глобальний характер.
Канцерогени — хімічні, фізичні і біологічні агенти природного і антропогенного походження — здатні за певних умов індукувати рак тварин і людини.
Фізичні чинники. До фізичних канцерогенних чинників відносяться: корпускулярні (а- і в-) випромінювання, іонізуючі випромінювання (рентгенівські і г-промені), не іонізуючі ультрафіолетові промені і, нарешті, хімічно інертні речовини (або що вважаються такими).
Іонізуюче випромінювання — один з найвідоміших екзогенних чинників канцерогенезу. Воно викликає появу пухлини, причину якої легко пов'язати з його дією, наприклад, коли пухлина з'являється в опроміненій анатомічній зоні. Частота таких злоякісних пухлин зростає у міру підвищення дози опромінювання і не зменшується при захисті інших ділянок тіла. Найбільш яскравий приклад — пухлини шкіри, що зустрічаються у рентгенологів. Помірне, але тривале сумарне опромінювання, якому піддавалася людина, може бути причиною різних видів лейкозу. Відома відносно висока частота їх серед рентгенологів, а також жителів Нагасакі і Хіросіми, що пережили атомне бомбардування, у хворих, що лікувалися опромінюванням хребта з приводу ревматизму, у дітей, опромінених в утробі матери при рентгенографії тазу. При дослідженні випадків злоякісних захворювань, викликаних пренатальним опромінюванням, виявилася лінійна залежність між дозою і частотою виникнення пухлин. Залишається спірним питання про існування порогової дози опромінювання, нижче за яку не виявляється канцерогенна дія.
Ультрафіолетові промені мають свої особливості. Вони не володіють іонізуючими властивостями і проникають в тканини організму не глибше чим на 2 мм (їх проникаюча здатність нижче проникаючої здатності іонізуючого випромінювання, оскільки довжина їх хвилі значно більше). Не дивно, що найчастіше вони викликають рак шкіри. Відповідний комітет Національної академії наук США встановив роль дії ультрафіолетового сонячного випромінювання в 80% випадків карцином і 40% випадків меланом шкіри. У експерименті вдавалося викликати пухлини в місці імплантації різних речовин.
Хімічні чинники. Понад 1000 хімічних речовин, які людина поглинає з їжею, питтям, вдихає і в яких він купається, можуть бути потенційними канцерогенами. Вони належать до всіх хімічних класів. Першими були вивчені поліциклічні вуглеводні, після того, як в 1775 р. з'ясувалася роль сажі (а значить, і кам'яновугільної смоли, яку вона містить) у виникненні раки мошонки у сажотрусів.
140 років через японські учені Ямагива і Ішикава викликали у кроликів рак шкіри при змазуванні її кам'яновугільною смолою. У тварин, як і у людини, пухлини з'являються тільки через місяці і роки після контакту з канцерогенною речовиною (так званий латентний період). З'ясувалося, що смола, що отримується з вугілля, нагрітого до 1200° З, не канцерогенна, а з вугілля, нагрітого тільки до 800° З, - сильний канцероген. Перегонка смоли при різних температурах дозволила виявити канцерогенність різних її фракцій. Так була виявлена причетність до канцерогенезу деяких вуглеводнів і виділений з кам'яновугільної смоли один з них — 3,4-бензпирен. Інші вуглеводні були синтезовані або виділені із смол, отриманих за допомогою синтезу. Складовою частиною канцерогенних вуглеводнів є антрацен, хімічна формула якого має в своєму складі три бензолові кільця. Дібензантрацен, як випливає з його назви, - похідний бензантрацена, один з сильних канцерогенних вуглеводнів. Існує цілий ряд канцерогенних вуглеводнів, визнаних сьогодні небезпечними для людини, використовуваних в промисловості і побуті, в онкологічних лабораторіях для відтворення різноманітних експериментальних пухлин. Найчастіше в лабораторії використовується метилхолантрен.
У експерименті як канцерогени удаються до деяких похідних хлора: четыреххлористому вуглецю, хлороформу, гептахлору, різним пестицидам (ДДТ і диальдрин). Відомо, що хлорвініл — мономер, використовуваний для виробництва пластмас, що є його полімером, - може викликати рак печінки у людини і щурів. Особливо небезпечні деякі похідні азоту; краще всього вивчені аміни. З кінця минулого століття відомо, що похідні ароматичного аміну — аніліну — викликають рак сечових шляхів, зокрема сечового міхура; в цьому відношенні найбільшу небезпеку представляє нафтиламин. Інше похідне аніліну — фенацетин, знеболюючий штучний препарат, — вивчається як вірогідний канцероген. Є окремі спостереження за онкологічними хворими, які широко користувалися фенацетином задовго до появи у них пухлин.
Зовсім недавно встановлено, що в шлунку щура відбувається абсорбція нітриту і деяких азотистих з'єднань (амінів і амидов), які, вступаючи в хімічну реакцію, утворюють нитрозамины — сильні канцерогени. Встановлена також канцерогенність синтетичних органічних сполук (використовуваних як протипухлинні препарати) і статевих гормонів. Серед канцерогенів опинився уретан, яким перестали користуватися в лікувальній меті, але все ще користуються в лабораторіях.
Відомі канцерогенні властивості органічних речовин рослинного походження (дубильних речовин і деяких алкалоїдів), ліків (як підозрює Армстронг, резерпин сприяє захворюванню раком молочної залози у жінок), речовин, що виділяються грибами (афлатоксин — продукт цвілі земляного горіха і ін.), з'єднань, що утворюються бактеріями (этионина, що виділяється кишковою паличкою) і ін. Навіть серед мінеральних речовин є канцерогени: азбест (на першому місці), хром, нікель, кадмій, свинець, олово, залізо, миш'як...
Комбіновані канцерогени. Канцерогени можуть складатися з активної речовини з доведеною або передбачуваною канцерогенністю або нести в організм інші речовини, що є вірогідними канцерогенами. При цьому утворюються канцерогенні суміші і комбінації.
Роль тютюну в появі деяких пухлин людини повністю доведена численними епідеміологічними дослідженнями, проведеними майже у всіх країнах. Найчастіше у курців виникає рак легені. Ризик появи цієї раки чітко коррелируется з кількістю викурюваних сигарет.
Відома безліч типів раки, виникнення яких може бути пов'язане з курінням тютюну. Вони зустрічаються з різною частотою. Як приклад комплексних канцерогенів можуть розглядатися алкогольні напої, оскільки вони не уживаються у вигляді чистого етилового спирту, а містять в собі продукти перегонки і добавки, серед яких є поліциклічні вуглеводні, нитрозамины, афлатоксины і так далі Крім того, спирт — чудовий розчинник деяких з цих речовин, а його роль в запаленні слизистої оболонки верхніх відділів травного тракту очевидна. Пухлини, що викликаються алкоголем, трохи менше поширені, чим що викликаються тютюном, але різновидів їх більше. Вірогідність виявлення раки у алкоголіків (в порівнянні з непитущими) в п'ять з гаком раз більше для пухлин рота, в 10 разів більше для пухлин гортані і в 17 разів — стравоходу. Підвищення канцерогенної риски, що викликається алкоголем, добре доведене для раки молочної і щитовидної залоз, меланом шкіри. Як підозрюють, алкоголь впливає на частоту виникнення раки шлунку, підшлункової і передміхурової залоз. Доведено, що у любителів пива підвищений ризик раки прямої кишки. Тільки зовсім недавно деякі епідеміологи на основі достовірних даних звинуватили каву в причетності до появи раки ряду локалізацій, зокрема сечового міхура.
На виникнення пухлин комплексну дію надають продукти живлення, в яких важко виділити відносний вплив різних якісних і кількісних чинників. Доведений зв'язок високої частоти раки, зокрема пухлин, не пов'язаних з травною системою, з ожирінням. Встановлена чітка кореляція між висококалорійною їжею і частотою раки молочної залози у жінок. Яскравим проявом даної закономірності може служити дуже висока частота раки цієї локалізації у американок і дуже низька у японок в порівнянні з середнім світовим рівнем. Щури, яким згодовували малокалорійний корм, в три рази рідше вражалися раком, чим щури, що знаходилися на рясному раціоні. Надлишок білків в кормі підвищує у щурів частоту пухлин, що викликаються хімічними канцерогенами, а мізерний, раціон зменшує у них частоту пухлин, нитрозаминами, що викликаються. Вплив такого раціону може бути часткове непрямим: білки, що містяться в нім, змінюють активність ферментів печінки, здатні перетворювати на канцерогени речовини, які такими не є, і наборовся.
Деякі складові частини білків (окремі амінокислоти) виконують особливу роль: так, добавка триптофана в їжу підвищує частоту раки печінки, нитрозамином, що викликається.
Біологічні чинники. Живі організми, а саме деякі гельмінти, бактерії і віруси, можуть грати провідну роль в появі раки. Шистосоми — найбільш драматичний приклад гельмінта, сприяючого розвитку злоякісних новоутворень. Вони викликають різні пошкодження внутрішніх органів залежно від виду паразита. Зокрема, в Єгипті спостерігається шистосомный цистит, який дуже часто ускладнюється раком сечового міхура. І дійсно, це найбільш частий вид злоякісної пухлини серед населення даної країни: від шистосоматоза і раки, що викликається ним, залежить поки що не високий (50 років) прогноз життя єгиптян. Кажучи про це, ми маємо на увазі той факт, що запалення проявляє себе як попередник раки. Запалення сечового міхура не єдиний випадок хронічного запалення, сприяючого раку. В принципі важливо виявити зв'язок між кількістю і співвідношенням різних бактерій в організмі або в деяких його анатомічних відділах, з одного боку, і частотою раки різних локалізацій, з іншою. Факти свідчать на користь можливого впливу стану кишкової флори на виникнення раки не тільки товстої кишки, але і молочної залози. Відмічений також зв'язок між інфекцією сечових шляхів і захворюваністю раком – шлунку. У першому випадку допускають, що кишкові бактерії виробляють канцерогени з харчових речовин і складових частин жовчі, в другому випадку можлива участь мікробів в перетворенні нітратів в нитрозамины.
Екологічні спостереження послужили основою для вивчення радіаційного і хімічного канцерогенезу задовго до народження онкоэпидемиологии як медичної дисципліни. Тепер, через 75 років після того, як Боррелем була висловлена вірусна теорія канцерогенезу і пройшло понад 10 років з моменту активного її розвитку, епідеміологи все ще шукають доказу участі вірусів в канцерогенезі. Але вже сьогодні можна стверджувати, що деякі віруси, виділені від тварини або людини, є сильними канцерогенами для тварин, у яких вони виявлені. Віруси грають основну роль в спонтанному канцерогенезі цих і інших видів тварин. Розумно припустити, що віруси грають не менш важливу роль в канцерогенезі у людини. Поки ж достатньо достовірні дані про вірусний канцерогенез стосуються тільки експериментів. Випередження експериментальними дослідженнями клінічних в цій області вельми значно: відкриття першого відомого канцерогенного вірусу, вірусу лейкозу курнув, відбулося на початку поточного сторіччя. Одні віруси викликають появу спонтанних пухлин в більшості випадків злоякісних, інші — доброякісних пухлин (фібром і папілом). Одні пухлини утворюються у тварин, не підданих відбору по генетичних ознаках, інші головним чином у тваринних чистих ліній (рак молочної залози і лейкоз у мишей). Дія вірусів була розкрита дією ультрафильтрата пухлинної тканини на здорових тваринах. Пухлини з'являються тільки після більш менш тривалого латентного періоду. В деяких випадках канцерогенний ефект не залежить від віку реципієнта. У інших же, зокрема при спонтанному лейкозі мишей, розвиток злоякісного захворювання відбувається тільки при зараженні негайно після народження особини. Це експериментальне спостереження дозволило припустити, що існують два типи передачі канцерогенних вірусів: горизонтальна, здійснювана різними носіями і що діє незалежно від віку реципієнта, і вертикальна, здійснювана тільки у новонароджених або плоду; її носії сперма, яйцеклітина і молоко.
Як відомо, існують ДНК- і РНК – віруси. ДНК-вірус здатна або зруйнувати, або змінити клітки, які вони інфікують. Як правило, в змінених клітках ділення вірусу припиняється (тобто ці клітки не проводять інфекційних вірусів), а наявність вірусної інформації виражається тільки появою нових антигенів (кодованих вірусним геномом). Різна канцерогенна ДНК-вірус істотно відрізняється один від одного морфологічними ознаками.
РНК-вирусы поводяться по-різному: вони не руйнують клітку; деякі з них трансформують її. Цикл розмноження вірусу може продовжуватися безперервно і незалежно від клітинного ділення і закінчуватися утворенням нових вірусних частинок. Канцерогенність вірусу РНК зазвичай досить слабка. Вона яскравіше виявляється при індукції сарком і слабкіше — при розвитку лейкозу. Серед ДНК- і РНК-вирусов є і такі, які не можуть закінчувати цикл свого розвитку утворенням повних вірусних частинок. Їх називають дефектними. Деякі РНК-вирусы, що зокрема викликають саркому, потребують допоміжного вірусу, званому «хелпер», а саме вірусі лейкозу, який забезпечує вірус саркоми поряд молекул для побудови мембрани.
Серед ДНК-вірусу, виділеної від людини, «кандидат номер один» в групу вірусів, що мають етіологічне значення в канцерогенезі, вірус Епштейна — Барр (ЕБВ), який належить до сімейства вірусів герпесу. Якщо припустити, що ЕБВ є канцерогенним для людини, факт його повсюдного розповсюдження свідчить про те, що для виникнення пухлини під впливом цього вірусу потрібні додаткові умови. У Африці географічний збіг зон малярії і лімфоми Беркитта дозволив припустити, що додатковим чинником є малярія.
Серед іншої ДНК-вірусу, виділеної від людини, заслуговують розгляду три групи: аденовіруси; віруси, що викликають бородавки, і вірус гепатиту В, або «австралійський антиген». Відомо, що аденовіруси людини викликають рак у хом'яків, але пошуки самих вірусів або антитіл проти них, проведені у онкологічних хворих, не увінчалися успіхом. Є віруси, відомі тим, що вони викликають появу бородавок, але немає даних, що дозволяють з упевненістю говорити про те, що вони викликають рак у людей. Нарешті, вірус гепатиту В був запідозрений в тому, що він викликає рак печінки тільки у жителів Африки (де його антиген частіше виявляється у хворих раком печінки). Підозрюють також, що вірус викликає і лейкоз (як вважають, лейкоз пов'язаний із стійкістю вірусу, що передається найчастіше при переливанні крові). Тут необхідно нагадати, що вірусний гепатит може ускладнюватися хворобою кісткового мозку — аплазією, тобто зникненням кліток — продуцентів крові; ця хвороба сама по собі іноді ускладнюється лейкозом.
Деякі РНК-вирусы, виділені від людини, звернули на себе увагу епідеміологів і біологів. Їх цікавить, наприклад, питання: чи не підвищує вірус грипу, коли він інфікує плід в утробі матери, ризик, що діти захворіють лейкозом? Особливо наполегливо досліджуються канцерогенні РНК-вирусы при пухлинах людини, відповідних трем типам злоякісних захворювань, що викликаються вірусами у мишей і інших тварин: лейкозу, саркомам і раку молочної залози.
Розглянуті вище елементи (протоонкогены, клітинні онкогены, антионкогены, втрата гетерозиготности по генам-супрессорам, асоціації з генетичними маркерами) і чинники канцерогенезу не вичерпують всій многокомпонентности генетичного нахилу до раку і многофакторности причин пухлинного процесу. Можна визначити, щонайменше ще дві групи спадкових характеристик індивіда, що мають відношення до канцерогенезу
По-перше, процеси репарації ДНК. У підтримці генетичного гомеостазу ДНК і стабільності генетичних структур клітки, а саме вони визначають нормальну поведінку клітки, істотну роль грають репаративные процеси. Організм людини має в своєму розпорядженні унікальні можливості репарації що виникають спонтанно або під впливом зовнішніх чинників пошкоджень ДНК, ведучих до мутацій (темновая, эксцизионная репарація, позаплановий синтез ДНК). Спадкові аномалії в системах репарації ДНК ведуть до злоякісних новоутворень (пігментна ксеродерма, спадковий неполипозный колоректальный рак, атаксия-телеангиэктазия і ін.).
По-друге, метаболізм канцерогенів в організмі визначається біохімічними системами, багато хто з яких представлений генетично поліморфними формами. Поліморфізм ферментів (эстеразы, оксигенази, изоформа цитохрома Р450), що активують ксенобіотики, і детоксицирующих ферментів (різні трансферазы) визначає індивідуальну чутливість до канцерогенних дій.
Таким чином, медико-генетическое консультування сім'ї із спадковою схильністю до злоякісних новоутворень — складне завдання. Потрібні спеціальна підготовка врачей- генетиків в питаннях генетичних основ канцерогенезу і хороша лабораторна база. Тільки таке поєднання дозволить правильно виявити схильність членів сім'ї, визначити необхідність їх диспансерного спостереження і характер профілактичних заходів.