

4.4.1. Ізолюючі технології виробництва стерильних лікарських засобів
Вирішення проблем пов’язаних з присутністю персоналу дозволяють ізолюючі технології. Основним джерелом контамінації у виробничому приміщенні є персонал. Доцільно розглянути можливі джерела забруднення повітря чистих приміщень контамінантами різної походження.
Джерела мікрозабруднень. Оточуюче нас повітря містить велику кількість як живих (життєздатних), так і неживих частинок, що відрізняються по своїй природі і розмірам.
Мікрозабруднення виділяються персоналом, огороджуючими конструкціями, обладнанням, надходять у чисте приміщення з оточуючого простору. В чистому приміщенні 70-80 % присутніх мікрозабруднень приходиться на людину, 15-20% - на обладнання, 5-10% - на навколишнє середовище (рис. 4.1.)
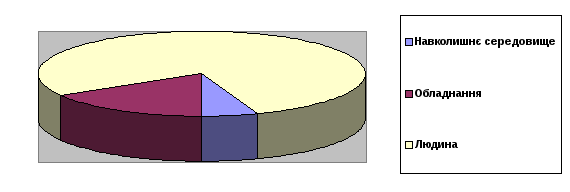
Рис. 4.1. Джерела мікрозабруднень.
З аналізу природи мікрозабруднень у повітрі і характерних для них розмірів видно, що проблема чистих приміщень носить комплексний характер. Недостатньо створити власно чисте приміщення, що забезпечує потрібний клас чистоти за відсутності технологічного устаткування і персоналу. Потрібно одночасно застосовувати устаткування, що виділяє мінімум забруднень або не виділяє їх узагалі, одягти людей у одяг, що не пропускає частинки, які виділяє людина під час фізичної активності та ін.
Остаточно з’ясовано, що основним джерелом забруднень у чистому приміщенні є людина. Це пояснюється структурою шкіри людини і динамікою її зміни. Зовнішній покрив шкіри людини має безлічі пластинок (лусочок) розміром приблизно в одиниці і десятки мікрометрів. Вони постійно відокремлюються з поверхні шкіри таким чином, що кожні кілька днів зовнішній шкірний покров цілком оновлюється. Відокремившись, вони розділяються на більше дрібні частки. У спокійному, нерухомому стані людина виділяє у хвилину приблизно 200 тис. часток розміром 0,5 мкм і більше. Навіть невеликі рухи і тертя тіла об одяг приводять до різкого збільшення викиду частинок до декількох мільйонів у хвилину. При інтенсивному русі людина виділяє приблизно 10 млн. частинок у хвилину. У середньому людина виділяє близько 3,5 кг частинок за рік або 10 г у день.
При русі в чистих приміщеннях люди в халатах і лабораторному одязі виділяють у навколишнє середовище в середньому:
два мільйони частинок розміром 0,5 мкм і більше,
триста тисяч частинок розміром 5 мкм і більше,
160 частинок, але з мікроорганізмами.
Джерелами інтенсивного виділення часток є ніс і рот людини. Інтенсивність виділення різко зростає при розмові, особливо під час голосної мови і лементу. Частинки, відділившись від людини, підхоплюються постійним природним конвекційним потоком повітря, що рухається нагору і весь час оточує людину. Далі вони поширюються по всьому об’єму приміщення й осідають на устаткуванні, матеріалах, продукті, огороджуючих конструкціях, інших працівниках і т.д. При цьому частинки можуть бути носіями мікроорганізмів.
Відповідно до статистики на 1000 зважених часток приходиться орієнтовно один мікроорганізм. У стандарті Національного агенції по дослідженню космосу США (NАSА) NНВ 5340 наводиться зразкове співвідношення між числом частинок і мікроорганізмів у повітрі (табл. 4.7.). Звичайно, з-за множинності факторів, що впливають на мікробне забруднення, ці дані носять наближений, ймовірний характер, але проте дають уявлення про взаємозв’язок між цими ключовими параметрами. Націленість одержати хоча б орієнтований зв’язок між числом частинок і числом мікроорганізмів у повітрі було викликано практичними розуміннями. Рахування частинок у повітрі проводиться швидко і легко. Це зручно при атестації чистих приміщень і поточному контролі. Аналіз же мікробного забруднення вимагає часу і затрат.
Таблиця 4.7. Зв’язок між числом частинок і числом мікроорганізмів у повітрі по стандарту NАSА NНВ 5340
|
Клас чистого приміщення по стандарту США 209 D |
Частинки |
Бактерії |
||
|
Діаметр, мкм |
Кількість, 1куб. фут (л) |
Зважені, 1куб. фут (л) |
Осаджені, 1кв. фут/нед. (1кв м/нед.). |
|
|
100 |
>0,5 |
<100 (<0,35) |
<0,1 (<0,0035) |
1200 (12900) |
|
10000 |
>0,5 |
<10000 (<350) |
<0.5 (<0.0176)
|
6000 (64600) |
|
>5,0 |
<65 (<2,3) |
|||
|
100000 |
>5,0 |
<100000 (<3500) |
<2.5 (<0.0884) |
30000 (323000) |
|
>0,5 |
<700 (<25) |
|||
Серйозним кроком вперед у забезпеченні стерильності продукту, підвищенні ефективності чистих приміщень і чистих зон і зниженні пов’язаних з ними витрат є застосування ізолюючої технології і технології видування-наповнення-герметизація.
Суть ізолюючої або бар’єрної технології полягає у фізичній ізоляції робочої зони від навколишнього простору за рахунок застосування герметичного ізолятора. Ця технологія широко впроваджується у виробництві стерильних препаратів. Правилами GМР ЄС з 1997р. установлено, що для випадку асептичного виробництва простір, що оточує ізолятор, повинен відповідати принаймні зоні D. Це істотно більше проста умова, ніж вимоги до чистоти в звичайній технології чистих приміщень. Воно дозволяє значно знизити витрати на будівництво і експлуатацію чистих приміщень.
Використання ізолюючої технології знімає необхідність присутності людини у виробничих зонах, у результаті чого значно знижується ризик контамінації продукції, виробленої в асептичних умовах, із навколишнього середовища. Існує багато типів ізоляторів і передавальних пристроїв. Ізолятор і оточуюче його середовище мають бути сконструйовані таким чином, щоб у відповідній зоні забезпечувалася необхідна якість повітря. Конструкції передавальних пристроїв можуть варіювати від пристроїв з одинарними або подвійними дверима до повністю герметизованих систем, включаючи стерилізацію.
Передача матеріалів усередину і назовні пристрою є одним із найсерйозніших потенційних джерел контамінації. Звичайно простір усередині ізолятора є обмеженою зоною для ведення операцій, що потребують мінімального ризику контамінації або його відсутності, хоча визнано, що в робочій зоні всіх таких пристроїв може бути відсутній ламінарний потік повітря. Чистоту цього простору необхідно контролювати; виробництво в асептичних умовах потребує принаймні клас чистоти D.
Експлуатація ізоляторів може бути почата тільки після проведення відповідної валідації. Валідація повинна враховувати всі критичні фактори ізолюючої технології, наприклад, якість повітря усередині та зовні (навколишнього простору) ізолятора, санітарну обробку ізолятора, процеси передачі та цілісність ізолятора. Необхідно постійно проводити контроль, який включає часті випробування герметичності ізолятора і його вузлів.