

Журнал неврологии и психиатрии / 2006 / NEV_2006_10_11
.pdfОБЗОРЫ
Артериальная гипертония и головной мозг
В.И. СКВОРЦОВА, А.Ю. БОЦИНА, К.В. КОЛЬЦОВА, И.А. ПЛАТОНОВА, К.И. ПОЧИГАЕВА, К.В. СОКОЛОВ, Т.В. ТВОРОГОВА
Arterial hypertension and the brain
V.I. SKVORTSOVA, A.YU. BOTSINA, K.V. KOLTSOVA, I.A. PLATONOVA, K.I. POCHIGAEVA, K.V. SOKOLOV, T.V. TVOROGOVA
Кафедра фундаментальной и клинической неврологии Российского государственного медицинского университета, Москва
Артериальная гипертония (АГ) в настоящее время признана пандемией, определяющей структуру заболеваемости в большинстве стран мира, в том числе в Российской Федерации. По данным обследования репрезентативной выборки, стандартизованная по возрасту распространенность артериальной гипертонии в России составляет среди мужчин 39,2%, а среди женщин — 41,1%1.
Нервная, иммунная и эндокринная системы выполняют совместную функцию сохранения динамического гомеостаза в организме. Являясь компонентами единой системы, они взаимодействуют по принципу взаимной регуляции, осуществляемой нейромедиаторами, нейропептидами, цитокинами, трофическими факторами, гормонами через соответствующий рецепторный аппарат [21]. Взаиморегуляция нервной, эндокринной и иммунной систем определяет надежность их совместной деятельности. В то же время она создает риск развития функциональных расстройств общей системы при первичном нарушении какой-либо из подсистем. Такого рода расстройства можно определить как дизрегуляционную патологию, патогенез которой может быть связан с первично нервными, эндокринными и/или иммунными механизмами [10].
Рассматривая взаимоотношения артериальной гипертонии и морфофункционального состояния головного мозга, следует выделить 2 аспекта: участие головного мозга в формировании артериальной гипертонии и влияние артериальной гипертонии на состояние головного мозга.
Участие головного мозга в формировании артериальной гипертонии
В последние годы пристальное внимание исследователей направлено на изучение нейрогенных механизмов артериальной гипертонии. Известна роль высших вегетативных центров, локализованных в лимбико-ретикулярном комплексе, и симпатического отдела вегетативной (автономной) нервной системы в развитии артериальной гипертонии. Однако ранее изучались преимущественно периферические механизмы влияния симпатической нервной системы на сократительную способность сосудов и миокарда, опосредуемые через систему аминергической нейротрансмиссии (катехоламины) и адренергические рецепторы. Расположение катехоламинергических нейронов в головном мозге в настоящее время уточняется.
Норадренергические нервные клетки находятся только в узкой переднелатеральной зоне покрышки продолговатого мозга и моста (А1—А7). Самая большая группа норадренергических клеток А6 расположена в области голубого пятна (ядра) и включает почти половину всех норадренергиче- ских клеток (не более 1000). У взрослых голубое пятно вклю- чает нейромеланин-содержащие нервные клетки, форми-
© Коллектив авторов, 2006
Zh Nevrol Psikhiatr Im SS Korsakova 2006;106:10:68—78
рующие темно-синюю полоску длиной около 1 см в верхней части моста у дна четвертого желудочка. Ядро простирается вверх вплоть до нижних холмиков четверохолмия. Аксоны клеток голубого пятна многократно ветвятся, их адренергические окончания можно найти во многих отделах ЦНС. Волокна, идущие от этих групп норадренергиче- ских нервных клеток, или поднимаются к среднему мозгу, или нисходят к спинному мозгу. Кроме того, норадренерги- ческие клетки имеют связи с мозжечком. Норадренергиче- ские волокна разветвляются обширнее, чем дофаминерги- ческие. Близость норадренергических волокон к артериолам и капиллярам мозга поразительна, не случайно они играют важную роль в регуляции мозгового кровотока.
Адреналин (эпинефрин)-синтезирующие нейроны находятся только в продолговатом мозге, в узкой переднелатеральной области. Наибольшая группа клеток С1 лежит позади заднего оливного ядра, средняя группа клеток С2 — рядом с ядром одиночного пути, и группа клеток С3 — непосредственно под перивентрикулярным серым веществом. Эфферентные пути от С1—С3 идут к заднему (дорсальному) ядру блуждающего нерва, ядру одиночного пути, голубому пятну, перивентрикулярному серому веществу моста и среднего мозга, гипоталамусу и паравентрикулярным ядрам. Физиологические эксперименты показали, что группа клеток С1 является очень чувствительным вазопрессорным центром.
У катехоламинов описаны 4 главных типа рецепторов: α1, α2, β 1 è β2. Они различаются по реакции на различные агонисты или антагонисты и по постсинаптическим эффектам. Рецепторы α1 управляют кальциевыми каналами при помощи вторичного мессенджера инозитол-1,4,5-трифосфата (IP3) и при активации повышают внутриклеточную концентрацию ионов Ca2+. Рецепторы α2 ведут к уменьшению концентрации вторичного мессенджера цАМФ, что вызывает различные эффекты. Рецепторы β при помощи вторич- ного мессенджера цАМФ повышают проводимость мембран для ионов К+, генерируя тормозной постсинаптический потенциал.
Доказательства того, что стимуляция α2-адренергиче- ских рецепторов в гипоталамусе и области nucleus tractus solitarii (NTS) продолговатого мозга оказывает ингибирующий эффект на эфферентную симпатическую активность и приводит к вазодилатации, позволили предположить, что повышение АД может являться следствием пониженной чувствительности нейронов этих областей головного мозга или ослаблением синтеза в них норадреналина [6].
Экспериментальные работы последних лет установили участие молекулярных регуляторов, синтезируемых в нервной ткани, в том числе нейронального оксида азота (NO), в реализации нейрогенных механизмов формирования ар-
1 Данные Государственного научно-исследовательского центра профилактической медицины МЗ РФ, представленные в докладе ДАГ1. РМЖ 2000; 8: 8: 109: 318—346.
68 |
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 10, 2006 |

|
|
|
териальной гипертонии. Нейрональный оксид азота оказы- |
В сосудах активация Rho/Rho-киназного пути проис- |
|
вает ингибирующее влияние на симпатическую активность |
ходит под влиянием ангиотензина-II и эндотелина [54]. Та- |
|
как на центральном, так и на периферическом уровне. Внут- |
ким образом, система Rho/Rho-киназы участвует и в реа- |
|
рижелудочковое введение неселективного ингибитора син- |
лизации сосудистого воспаления и гипертрофического ре- |
|
теза оксида азота, блокирующего экспрессию и нейрональ- |
моделирования сосудистой стенки в патогенезе артериаль- |
|
ной (nNOS), и эндотелиальной (eNOS) NO-синтазы, ак- |
ной гипертонии. |
|
тивирует симпатическую нервную систему, вызывая досто- |
Одним из независимых факторов риска развития эндо- |
|
верное повышение артериального давления и частоты сер- |
телиальной дисфункции в рамках артериальной гипертонии |
|
дечных сокращений [89]. Введение ингибиторов синтеза NO |
является возраст [99]. Известно, что с возрастом активность |
|
(L-NMMA, L-NAME) в область nucleus tractus solitarii, вос- |
симпатической нервной системы в ряде периферических |
|
принимающего афферентные импульсы от баро- и хеморе- |
органов, включая сердце, существенно увеличивается [91]. |
|
цепторов артерий, также вызывает повышение АД и ЧСС |
Таким образом, возраст-ассоциированная активация сим- |
|
[33]. Таким образом, очевидно, что нейрональный NO не- |
патической нервной системы может объяснять увеличение |
|
посредственно воздействует на NTS и тем самым снижает |
риска развития артериальной гипертонии при взрослении и |
|
активность симпатической нервной системы. |
старении. |
|
Интересно, что введение селективного ингибитора ин- |
Безусловно, патология головного мозга вследствие его |
|
дуцибельной NO-синтазы (iNOS) аминогуанидина резко |
ишемического, травматического, неопластического и др. |
|
повышает уровень АД у крыс со спонтанной гипертонией |
повреждений может изменять реализацию АД-регулирую- |
|
только при условии предшествующего хронического тор- |
щих функций. Так, повреждение или дисфункция гипота- |
|
можения синтеза NO путем подавления экспрессии эндо- |
ламуса и миндалевидных ядер может проявляться наруше- |
|
телиальной и нейрональной NO-синтаз [84]. Вероятно, у |
нием обмена серотонина и пролактина, изменением синте- |
|
животных с хроническим торможением синтеза оксида азо- |
за морфиноподобных нейропептидов, активацией образо- |
|
та iNOS является единственным источником его продук- |
вания эндотелина, вазопрессина и дигиталисподобного на- |
|
ции. Однако синтезируемого с помощью iNOS уровня окси- |
трийуретического фактора в супраоптическом и паравен- |
|
да азота по-видимому недостаточно для эффективной вазо- |
трикулярных ядрах, что вносит существенный вклад в диз- |
|
дилатации. |
регуляцию системного АД и формирование артериальной |
|
Важно отметить, что у крыс со спонтанной артериаль- |
гипертонии. Изменения состояния норадренергических ней- |
|
ной гипертонией и предварительным хроническим тормо- |
ронов (А1—А2 групп) и адренергических нейронов (С1 груп- |
|
жением синтеза NO введение блокатора активности симпа- |
пы), расположенных в латеральных отделах продолговатого |
|
тической нервной системы пентолиниума вызывало досто- |
мозга и являющихся чрезвычайно чувствительным вазопрес- |
|
верно более значимое снижение системного АД, чем при |
сорным центром, также могут обусловить значительные |
|
применении ингибитора АПФ каптоприла, что косвенно |
колебания уровня АД. Установлено, что активация или тор- |
|
может свидетельствовать о доминирующем участии симпа- |
можение I1-имидазолиновых рецепторов, встроенных в мем- |
|
тической нервной системы в формировании артериальной |
браны этих нейронов, вызывает соответственно пониже- |
|
гипертонии по сравнению с участием ренин-ангиотензино- |
ние или повышение системного АД [20]. |
|
вой системы. |
Влияние артериальной гипертонии на состояние |
|
В процессах активации симпатической нервной сис- |
||
головного мозга |
||
темы и развития артериальной гипертонии особую роль |
||
играют функционирующие в нейронах головного мозга ки- |
Чрезвычайно важной проблемой является влияние по- |
|
назные сигнальные системы. Хроническое ингибирование |
||
вышенного артериального давления на морфофункциональ- |
||
синтеза NO приводит к активации ГТФазы Rho и ее ми- |
||
ное состояние головного мозга. Несмотря на то что более |
||
шени Rho-киназы. Rho оказывает непосредственное влия- |
||
высокий уровень АД ассоциируется с более высоким отно- |
||
ние на nucleus tractus solitarii, поскольку именно в нем, а |
||
сительным риском развития острого нарушения мозгового |
||
также в гиппокампе, мозжечке и коре больших полуша- |
||
кровообращения и инсульта, показано, что большую роль |
||
рий экспрессируется ген, кодирующий ее синтез. В норме |
||
в изменении состояния головного мозга играет усреднен- |
||
Rho выполняет множество разнообразных функций, в ча- |
||
ный уровень АД, нежели одномоментное его повышение. |
||
стности в головном мозге обеспечивает рост аксонов и |
||
Повышенное АД приводит к ускоренному развитию цереб- |
||
трофическую поддержку дендритов, формирующих пост- |
||
роваскулярных заболеваний и как следствие к формирова- |
||
синаптические мембраны большого количества синапсов; |
||
нию хронической ишемии головного мозга (по МКБ-10, |
||
участвует в регуляции экзоцитоза нейротрансмиттеров. Та- |
||
рубрика 167.8) или острых нарушений мозгового кровооб- |
||
ким образом, Rho/Rho-киназный сигнальный путь играет |
||
ращения (по МКБ-10, рубрика 160) [18]. В то же время осо- |
||
большую роль в реализации синаптической передачи. Тор- |
||
бую значимость имеет прямое действие артериальной ги- |
||
можение Rho/Rho-киназного пути в nucleus tractus solitarii |
||
пертонии на молекулярные, биохимические и клеточные |
||
приводит к снижению АД и ЧСС путем подавления сим- |
||
механизмы функционирования головного мозга, на микро- |
||
патической активности. При этом выраженность снижения |
||
циркуляторно-тканевые взаимоотношения. |
||
АД и ЧСС значительно превалирует у крыс со спонтанной |
||
Длительное существование артериальной гипертонии |
||
артериальной гипертонией по сравнению с контрольными |
||
включает основные механизмы развития хронического па- |
||
животными [51]. В настоящее время неизвестно, каким |
||
тологического (нейродегенеративного) процесса в ткани |
||
образом активация Rho/Rho-киназного пути в nucleus trac- |
||
головного мозга: хроническое воспаление, изменение про- |
||
tus solitarii приводит к изменению артериального давления |
||
ницаемости гематоэнцефалического барьера и аутоиммуни- |
||
и почему подобные изменения более выражены у крыс со |
||
зацию организма к собственным нейроспецифическим бел- |
||
спонтанной артериальной гипертонией. Возможно, инги- |
||
кам с последующим вторичным аутоиммунным поврежде- |
||
бирование Rho/Rho-киназного пути cвязано с возникно- |
||
нием головного мозга, митохондриальную дисфункцию и |
||
вением морфологических изменений в дендритах и аксо- |
||
оксидантный стресс, программированную клеточную смерть |
||
нах и, как следствие, с нарушением синаптической пере- |
||
(апоптоз) и дефицит трофических факторов. |
||
дачи афферентных вазоконстрикторных импульсов в nu- |
||
|
||
cleus tractus solitarii. Возможно также, что Rho-киназа во- |
Хроническое воспаление, изменения церебральной |
|
влечена в сигнальные пути, активированные ангиотензи- |
микроциркуляции и проницаемости |
|
ном-II, поскольку в центральной нервной системе он уча- |
гематоэнцефалического барьера |
|
ствует в реализации механизмов развития артериальной |
Исследования A. Siren и соавт. [93] показали, что дли- |
|
гипертонии [47]. |
тельная артериальная гипертония может вызывать актива- |
|
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 10, 2006 |
69 |

ОБЗОРЫ
цию внутриклеточных молекул адгезии (intercellular adhesion molecule ICAM-1) на эндотелиальных клетках и периваскулярное накопление лейкоцитов в ткани мозга, что отражает своеобразную предрасположенность к нарушению микроциркуляции у больных с артериальной гипертонией.
Иммуногистохимическое исследование сосудов крыс с гипертонией показало, что при депозиции фибриноидной субстанции в интиму сосудов мозга происходит активация экспрессии молекул ICAM-1, тромбоцит-эндотелиальных клеточных молекул адгезии (platelet-endothelial cell adhesion molecule (PECAM)-1), интерлейкина (IL)-1альфа, IL-6, IL-8
èфактора некроза опухолей (TNF)-α. С помощью сканирующей электронной микроскопии и трансмиссионной электронной микроскопии показана адгезия нейтрофилов, моноцитов и тромбоцитов к эндотелиальным клеткам, проникновение их через межклеточные контакты, расхождение контактов между эндотелиоцитами с депозицией фибриноидной субстанции в интиму, а также повреждение среднего слоя стенки артерий [40].
Являясь единственным иммунокомпетентным компартментом в центральной нервной системе, микроглия уча- ствует во всех реакциях церебральной ткани на повреждающее воздействие ишемии [107]. Хроническая ишемизация головного мозга, вызванная артериальной гипертонией, активирует микроглиальные клетки, приводя их в состояние готовности к фагоцитозу. По мнению R. Banati и соавт. [26], нейротоксическое влияние микроглиальных клеток осуществляется по трем основным механизмам: с помощью продукции прямых нейротоксических факторов; продукции микроглиальных факторов, запускающих патобиохимические каскады, которые приводят к клеточной смерти; а также с помощью индукции местного воспалительного ответа.
Установлено, что при нарушениях микроциркуляции и ишемизации ткани головного мозга микроглия начинает продуцировать широкий спектр токсичных для ткани мозга соединений: провоспалительные цитокины [24], лиганды для глутаматного NMDA рецепторного комплекса [43], протеазы, катепсин В, лизозимы, эйкозаноиды (в том числе тром-
боксан В2), супероксидный анион, нитроксид [34, 107] и, кроме того, инициирует цитотоксическое действие астроцитов [102]. Анализ перечня соединений, синтезируемых микроглиальными клетками, свидетельствует об активном
èсогласованном с другими клеточными пулами участии активированной микроглии во всех основных процессах глутамат-кальциевого каскада, поддерживающем глутаматную эксайтотоксичность, активацию внутриклеточных ферментов, свободнорадикальные реакции, перекисное окисление липидов. Однако наряду с этим микроглия выполняет
èспециализированные иммунные функции, индуцируя и поддерживая воспалительную реакцию в ткани головного мозга, что в конечном итоге ведет к отсроченным нейрональным потерям, изменениям микроциркуляции и гематоэнцефалического барьера [70, 106].
Диапедез лейкоцитов через эндотелий предшествует воспалению, а в месте воспалительных изменений в ткани головного мозга выявляются лейкоцитарные инфильтраты
èактивизированные периваскулярные макрофаги и микроглиальные клетки [37, 96]. После инфильтрации периваску-
лярной ткани активными лейкоцитами происходит повышение уровня провоспалительных цитокинов — TNF-α, IL1β, IL-6, продуцируемых как клетками гуморального ряда, так и местными периваскулярными макрофагами и микроглиальными клетками, а также эндотелиальными клетками
èастроцитами [38, 62, 96]. Локальная продукция цитокинов играет важную роль в дальнейшей активации эффекторных иммунных клеток и продукции медиаторов воспаления, приводящих к повреждению нейронов [61]. Высвобождение медиаторов воспаления сопровождается повышенной агрегацией тромбоцитов и увеличением показателя соотношения уровней тромбоксана А2 и простагландина I2,
что индуцирует дальнейшие нарушения микроциркуляции. Фактор агрегации тромбоцитов, являющийся провоспалительным медиатором, стимулирует лейкоциты и эндотелиальные клетки [69, 101].
После активации воспалительными медиаторами эндотелиальные клетки быстро вырабатывают адгезивные молекулы (например, Р-селектин — GMP-140), которыe накапливаются и вызывают дополнительное высвобождение ИЛ-1, TNF-α и фактора активации тромбоцитов. Полиморф- но-ядерные лейкоциты в циркулирующей крови также активируются адгезивными молекулами (Sialyl-Lewis X, Lewis X и CD11/18), расположенными на их поверхности. Под влиянием ИЛ-8 и других хемоаттрактантов лейкоциты притягиваются к ишемизированной зоне, скапливаются в ней вокруг микрососудов (преимущественно венул), на эндотелиальных клетках. Адгезия лейкоцитов к эндотелию опосредуется связывающими адгезивными молекулами (CD11/18 и ICAM-1) и может запускать активную систему генерирования свободных радикалов в эндотелиальных клетках (например, ксантиноксидазную), способную разрушить все перечисленные клеточные элементы [77]. Высвобождаясь из нейтрофилов, моноцитов и эндотелиальных клеток, воспалительные медиаторы (ИЛ-1, TNF-α, фактор активации тромбоцитов) продолжают разрушительные процессы. Под дейтвием TNF-α эндотелиальные клетки приобретают гемостатические прокоагулянтные свойства [78].
Молекулы клеточной адгезии участвуют в воспалительных реакциях, связывая активированные лейкоциты и опосредуя их роллинг и миграцию через эндотелий в окружающие ткани [68]. В условиях воспаления экпрессия этих молекул повышается под воздействием продуцируемых провоспалительных цитокинов, в частности интерферона (ИФ)-γ, TNF-α и интерлейкинов (IL-1β, IL-6) [30, 40, 92].
Многими исследователями была показана связь между повышением экспрессии молекул адгезии и повышенными уровнями циркулирующего ангиотензина II. В экспериментах на крысах со спонтанной артериальной гипертонией было показано, что на фоне постоянной гипертонии (уровень АД>220 мм рт.ст.) повышенная экспрессия ICAM-1 на эндотелии сосудов головного мозга снижалась при применении у животных ингибиторов АПФ и особенно при применении антагонистов рецепторов к ангиотензину II [50, 100]. У животных, которым вводили ангиотензин II, наблюдалось повышение артериального давления и повышение экспрессии ICAM-1, которое предотвращалось введением антагониста ангиотензина II лозартана. В то же время у животных, которым вводили фенилэфрин, также повышалось артериальное давление, однако повышения экспрессии ICAM не наблюдалось [55].
Установлено, что повышение экспрессии ICAM связано с активацией ядерного фактора NF-kappaB [35, 76]. В работе D. Muller и соавт. на трансгенных крысах, экспрессирующих человеческий ренин и ангиотензин, была продемонстрирована связь между повышенным уровнем NF-kap- paB и активацией рецепторов к ангиотензину [75]. В то же время торможение экспрессии NF-kappaB снижало экспрессию iNOS и явления воспаления в тканях у крыс. Повышенная экспрессия NF-kappaB также была ассоциирована с молекулами, имеющими окислительную активность [35], и эндотелином [76]. В эксперименте на эндотелиальных клетках пупочной вены было продемонстрировано, что активность NF-kappaB, вызванная окисленными липопротеинами низкой плотности или TNF-α, предотвращается ингибитором АПФ зофеноприлом, имеющим антиоксидантные свойства. В то же время энаприлат, не имеющий таких свойств, не ингибировал NF-kappaB и не предотвращал повышенной экспрессии ICAM-1, VCAM-1 и Е-selectin [76]. Интересно, что активированный NF-kappaB активирует синтез TNF-α, ИЛ-1β и ИЛ-6, которые в свою очередь повышают уровень NF-kappaB, таким образом создавая порочный круг [53].
70 |
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 10, 2006 |

|
|
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 10, 2006 |
71 |

ОБЗОРЫ
Рис. 1. Схема аутоиммунного повреждения головного мозга.
развитии деструктивных изменений [18]. Проникающие в ткань мозга антитела распространяются в межклеточном пространстве и далее транспортируются аксональным током в различные клеточные структуры, инактивируя соответствующие нейроантигены, что в конечном итоге приводит к усилению повреждения нервной ткани. Аутоантитела оказывают влияние как на метаболизм нервных клеток, так
èна формирование специфических функций нервной системы в целом [7, 11]. Таким образом, происходит развитие аутоиммунной агрессии, с механизмами которой связано хроническое и прогредиентное течение патологического процесса в центральной нервной системе. Аутоиммунные реакции являются важным патогенетическим звеном гипертонической энцефалопатии и энцефалопатий другой природы, при этом аутоантитела к нейроспецифическим белкам (как и сами нейроспецифические антигены) могут служить уникальными маркерами патологического состояния ткани головного мозга [1, 41, 48].
Нами было проведено клинико-иммунологическое исследование 40 больных с хронической ишемизацией головного мозга (дисциркуляторная энцефалопатия), не имевших в анамнезе инсультов (в том числе «немых», по результатам магнитно-резонансной томографии головного мозга). Определение в сыворотке крови аутоантител к фенцикли- дин-связывающему мембранному белку глутаматных NMDA рецепторов показало, что при близком к норме среднем по группе показателе титра (1,8±0,25 нг/мл) более чем у 40% больных выявляется его повышение до 2,5—4,0 нг/мл [5]. Детальный клинический анализ свидетельствовал о том, что все пациенты с повышенным титром аутоантител длительно страдали высокой артериальной гипертонией («рабочее» систолическое артериальное давление выше 180 мм рт.ст.) с повторными гипертоническими кризами. Учитывая влияние артериального давления на проницаемость ГЭБ представляется закономерной связь между выраженностью процессов аутоиммунизации к белковым компонентам нейрональных мембран и повреждающим действием прогредиентно развивающейся ишемии.
Для подтверждения «доинсультного генеза» аутоиммунных реакций к структурным нейроспецифическим белкам
было проведено исследование уровня аутоантител к фактору роста нервов (NGF), белку нейрональных мембран S100β
èосновному белку миелина (ОБМ) в сыворотке крови 40 больных с разными стадиями хронической ишемической болезни мозга (дисциркуляторной энцефалопатией 1-й — 3-й стадий) [4, 17]. У всех наблюдаемых больных дисциркуляторная энцефалопатия развивалась на фоне артериаль-
ной гипертонии, которая у 60% больных носила стойкий характер, а у 40% проявлялась в виде отдельных кризов, и атеросклероза, который в 10% случаев проявлялся гемодинамически значимыми стенозами или окклюзиями магистральных артерий головы. Наблюдение за больными осуществляли на протяжении 18 мес с шестикратным повторением клинико-иммунологических исследований [15].
Исследование в сыворотке крови аутоантител к NGF ни у одного наблюдаемого больного не определило значи- мого повышения титра. В то же время были выявлены существенные различия в показателях титра аутоантител к структурным белкам нервной ткани S100β и ОБМ. Близкие
êнорме (1:4, 1:8) титры аутоантител к S100β были выявле-
ны у 20% больных, к ОБМ — у 25%. Умеренно повышенные (1:16 — 1:64) титры к S100β и ОБМ регистрировали в 60 и 22,5% случаев соответственно; высокие (1:128, 1:256) титры — в 20 и 52,5%. Повторение контрольного исследования через 2 нед показало, что лишь у 2 наблюдаемых больных отмечались незначительные колебания титров антител (не более чем на 1 титр), что позволило считать выявленные изменения относительно стабильными.
Проведение корреляционного клинико-иммунологиче- ского анализа не выявило взаимосвязи уровней аутоантител к нейроспецифическим белкам в сыворотке крови с клиническими проявлениями и стадией дисциркуляторной энцефалопатии, а также вариантом течения артериальной гипертонии (стабильное или кризовое), выраженностью морфологических изменений ткани головного мозга (по данным магнитно-резонансной томографии) (r<0,15). Повышение титра антител не сопровождалось клиническими
признаками ухудшения состояния больных. Сопоставление титров антител к S100β и ОБМ устано-
вило, что у большинства больных (48%) значения титра антител к ОБМ были выше уровня антител к S100β, у 30%
исследованных показатели титров были одинаковы, у 22%
— превалировали значения титра антител к S100β. Такое распределение отражает индивидуальный характер морфологических изменений в ткани головного мозга на фоне хронической ишемии, а также свидетельствует о преобладании у большинства больных с дисциркуляторной энцефалопатией поражения белого вещества мозга [4]. Вместе с тем проведенный сравнительный анализ иммунологических показателей с результатами магнитно-резонансной томографии головного мозга не выявил различий в титрах антител
êОБМ в зависимости от выраженности лейкоарейоза и других изменений белого вещества. По-видимому, методы нейровизуализации и иммунологические тесты отражают процессы, не сопоставимые по времени.
Были установлены определенные закономерности в
соотношении между титрами первичных и вторичных антител к S100β. При наличии нормального титра первичных антител (1:2—1:8) уровень антиидиотипов был значительно повышен (1:32—1:128). Наоборот, наиболее высокому уровню первичных антител (1:128—1:512), как правило, соответствовали нормальные титры антиидиотипов. С учетом предварительных данных, полученных при ишемическом инсульте, можно полагать, что высокий индекс соотношения первичных и вторичных аутоантител является критерием «остроты» аутоиммунных процессов и, следовательно, патологической проницаемости ГЭБ [4, 94].
Всем наблюдаемым пациентам с дисциркуляторной энцефалопатией проводили повторные курсы нейропротективной терапии препаратом глицин (сублингвально в суточной дозе 600 мг). Комплексное клиническое (неврологическое, нейропсихологическое) исследование показало улучшение общего состояния больных на фоне лечения: уменьшалась выраженность несистемного головокружения, повышались внимание, работоспособность, общая активность, в то же время микроочаговая неврологическая симптоматика оставалась прежней. Клинико-иммунологический анализ свидетельствовал о том, что только через несколько
72 |
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 10, 2006 |

Рис. 2. Динамика титров антител к нейроспецифическим белкам в сыворотке крови больных с хронической ишемией головного мозга.
По оси ординат — титр антител, по оси абсцисс — период обследования. 1 — антитела к основному белку миелина, 2 — антитела к белку S100b, 3 — антиидиотипы к белку S100b.
недель после проведения первого курса терапии глицином у пациентов было выявлено значительное снижение титра первичных и вторичных аутоантител к нейроспецифическим белкам: у 65% — до 1:2 — 1:8, что соответствовало норме; у 35% — до 1:16 (рис. 2). Проведение повторного курса нейропротекции привело к дальнейшему снижению титров аутоантител, однако через 6 мес после прекращения приема глицина вновь проявлялась умеренная тенденция к нарастанию уровня антител.
Необходимо отметить, что у 3 пациентов за время наблюдения были зарегистрированы острые нарушения мозгового кровообращения, имевшие разное течение и очаговый морфологический дефект. У двух из них нарушения мозгового кровообращения развились на фоне повторного курса глицина и протекали как транзиторные ишемические атаки, без формирования очагового морфологического дефекта на магнитно-резонансной томограмме головного мозга. В то же время у третьего больного нарушение мозгового кровообращения возникло через 4 мес после окончания повторного курса глицина и характеризовалось тяжелым течением и обширным корково-подкорковым ишемическим
АРТЕРИАЛЬНАЯ ГИПЕРТОНИЯ И ГОЛОВНОЙ МОЗГ
поражением. Эти различия подчеркивают роль нейропротекции в торможении аутоагрессии к нейроспецифическим белкам и профилактике грубых морфологических повреждений ткани мозга на фоне нарушений его кровоснабжения [4].
Таким образом, у больных с хронической ишемизацией головного мозга на фоне артериальной гипертонии и атеросклероза развивается генерализованная аутоиммунизация к структурным компонентам нервной ткани. Аутоиммунные процессы принимают участие в формировании «фонового» сосудистого повреждения головного мозга (энцефалопатии) и предуготавливают церебральную ткань к развитию инфаркта в случае острого снижения мозгового кровотока. Курсы нейропротекторной терапии могут иметь профилактическую значимость для снижения темпов прогрессирования энцефалопатии и уменьшения риска развития крупноочаговых инфарктных изменений в головном мозге.
Митохондриальная дисфункция, реакция генома, программированная клеточная смерть, оксидантный стресс
Âнастоящее время очевидно, что контроль за смертью
èвыживанием клеток осуществляют множество молекулярных внутриклеточных регуляторов, жестко компартментализированных, и в то же время тесно взаимосвязанных внутриклеточной сигнальной системой (рис. 3).
Âпоследние десятилетия в качестве основной причи- ны первичного повышения АД рассматривается дефицит энергии на клеточном уровне. Источником нарушения энергетического обмена тканей при эссенциальной артериальной гипертонии считают снижение энергообразовательной функции митохондрий вследствие нарушения структуры митохондриального аппарата клеток [12]. При первичной (эссенциальной) гипертонии можно говорить о существовании генетически детерминированных особенностей клеточных мембран, инициирующих возникновение недостаточности мембранной регуляции внутриклеточного кальция с накоплением в цитоплазме высоких концентраций свободных ионов кальция, особенно при дополнительных физиологических нагрузках. Стабильное повышение АД происходит по мере аккумуляции митохондриями избытка цитозольного кальция, последующего снижения их энергооб-
Рис. 3. Схема молекулярного контроля за смертью/выживанием клетки (объяснение в тексте).
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 10, 2006 |
73 |
ОБЗОРЫ
разовательной функции и развития в полном объеме нарушения ионотранспортной функции клеточных мембран. Процессы аккумуляции избытка кальция и синтез АТФ в митохондриях тесно сопряжены, так как оба зависят от сохранности трансмембранного потенциала митохондрий [71]. Важным представляется факт, что при снижении трансмембранного потенциала не только уменьшается процесс синтеза АТФ, но и ускоряется гидролиз АТФ цитоплазмы, так как АТФ-синтаза начинает работать в обратном направлении для восстановления градиента протонов (феномен дыхательного контроля). Таким образом, повышенное системное АД можно рассматривать как системную особенность кровообращения, соответствующую сниженному уровню клеточной энергетики.
У животных с экспериментальной моделью первичной артериальной гипертонии получено доказательство того, что в основе данной патологии лежит процесс перестройки геномной ДНК [9, 13, 32]. При проведении экспериментальных работ установлено, что артериальная гипертония приводит к существенному перепрограммированию генного ответа клеток, изменению экспрессии генов раннего ответа c-fos, c-jun и c-myc [73, 88]. Система сигнальной трансдукции реагирует на любое повреждающее воздействие, в том числе на вызванную артериальной гипертонией церебральную ишемию, первой, принимая на себя функции регуляции всех молекулярных и клеточных событий, происходящих в ткани головного мозга. Центральным звеном в сигнальной цепи является рецептор-опосредованная генерация внутриклеточных вторичных мессенджеров, которые модулируют активность таких протеинов, как ферменты, рецепторы и ионные каналы. Все эти регуляторные протеины, включая рецепторы, участвуют в модуляции процессов нейронального повреждения и репарации. Основным механизмом передачи экстра- и интрацеллюлярных сигналов является фосфорилирование/дефосфорилирование протеинов с помощью ферментов протеинкиназ и фосфатаз, которые регулируются непосредственно рецепторной активацией или опосредованно — активированными вторичными мессенджерами (рис. 4). Перераспределение протеинкиназ на клеточных мембранах, вызванное ишемией, изменяет фосфо-
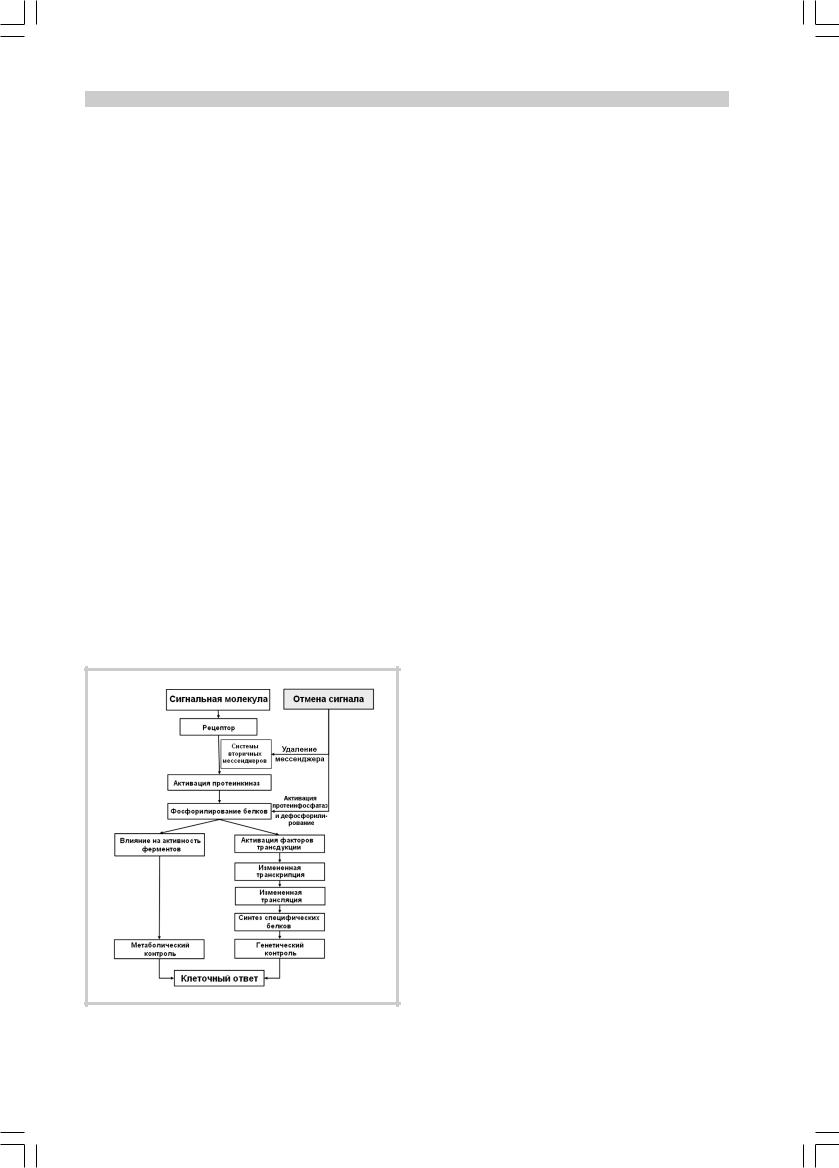
Рис. 4. Схема внутриклеточной сигнальной системы и передачи экстра- и интраклеточных сигналов.
рилирование рецепторов и протеинов ионных каналов. Состояние фосфорилированности регуляторных протеинов влияет на механизмы выживания и смерти клеток. Так, активация протеинкиназ стимулирует высвобождение нейротрансмиттеров, деятельность ионных каналов и рецепторов, что сопровождается нарушением активного ионного транспорта, истощением аденозинтрифосфата (АТФ). Дальнейшее фазное снижение активности протеинкиназ ведет к угнетению внутриклеточных сигнальных процессов, а следовательно, к уменьшению выраженности трофических и других жизнеобеспечивающих влияний на клетку, способствуя отсроченной нейрональной смерти.
Информация об изменении состояния мембранных структур и рецепторов, определяемая картиной перераспределения протеинкиназ на мембранах, передается с помощью систем вторичных мессенджеров (аденилатциклаз, протеинкиназ, фосфатаз) к ядру клетки, что является сигналом к «включению» единого триггерного молекулярного механизма, реализующего универсальный алгоритм ответа ткани мозга на повреждающее воздействие [74, 97]. Таким образом, микроциркуляторные изменения и хроническая ишемия активируют комплекс генетических программ, которые приводят к последовательной экспрессии большого числа генов.
Биохимические процессы генной экспрессии: первого этапа — транскрипции (биосинтеза молекул информационной, или матричной, РНК (мРНК) на матрице ДНК) и второго этапа — трансляции (процесса синтеза белка на основе кодовой последовательности нуклеотидов в мРНК) определяются доступностью АТФ и ГТФ и в целом сохранностью энергетического метаболизма [81]. Поэтому неудивительно, что первой реакцией ткани мозга на изменения микроциркуляции, вызванные артериальной гипертонией, является снижение синтеза мРНК и белков [108].
Вслед за активацией генов раннего реагирования и стресс-белков происходит активация генов, кодирующих молекулы универсальных регуляторов клеточных процессов, принимающих участие во всех основных механизмах «отсроченной» гибели клеток. На этом этапе происходит запуск синтеза провоспалительных цитокинов и молекул адгезии, которые индуцируют процессы воспаления в ткани головного мозга, нарушения микроциркуляции и гематоэнцефалического барьера [56, 66]; ферментов — синтазы оксида азота (iNOS) и циклооксигеназы-2 (COX-2), участвующих в механизмах оксидантного стресса [49, 79]. Экспрессия провоспалительных и прооксидантных факторов преимущественно происходит в нейтрофилах, эндотелиальных и микроглиальных клетках.
Наиболее значимым этапом нарушения деятельности клетки при падении трансмембранного потенциала митохондрий является нарушение проницаемости внутренней мембраны митохондрий с образованием комплексов «большой поры» и выходом ряда матриксных белков, многие из которых являются индукторами программированной клеточной гибели. Экспрессия генов раннего реагирования и синтез стресс-белков также непосредственно связаны с экспрессией генов индукторов апоптоза — одного из вариантов программированной клеточной смерти [67, 83].
Регуляция апоптоза в нервной системе осуществляется многочисленными сигнальными системами: с помощью модуляции факторов транскрипции (р53, АР-1, NF-kappaB) и ферментов, прямой активации генов раннего реагирования (с-jun, c-fos). Механизмы апоптоза запускаются тогда, когда вредное воздействие недостаточно сильно, чтобы вызвать некроз [105].
При артериальной гипертонии морфологические признаки апоптоза можно найти как в нейронах, так и в глиальных клетках головного мозга [13, 64, 67]. Установлено преимущественное апоптозное поражение олигодендроцитов [86]. Огромную значимость в развитии процесса апоптоза имеет недостаточность трофического обеспечения мозга.
74 |
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 10, 2006 |
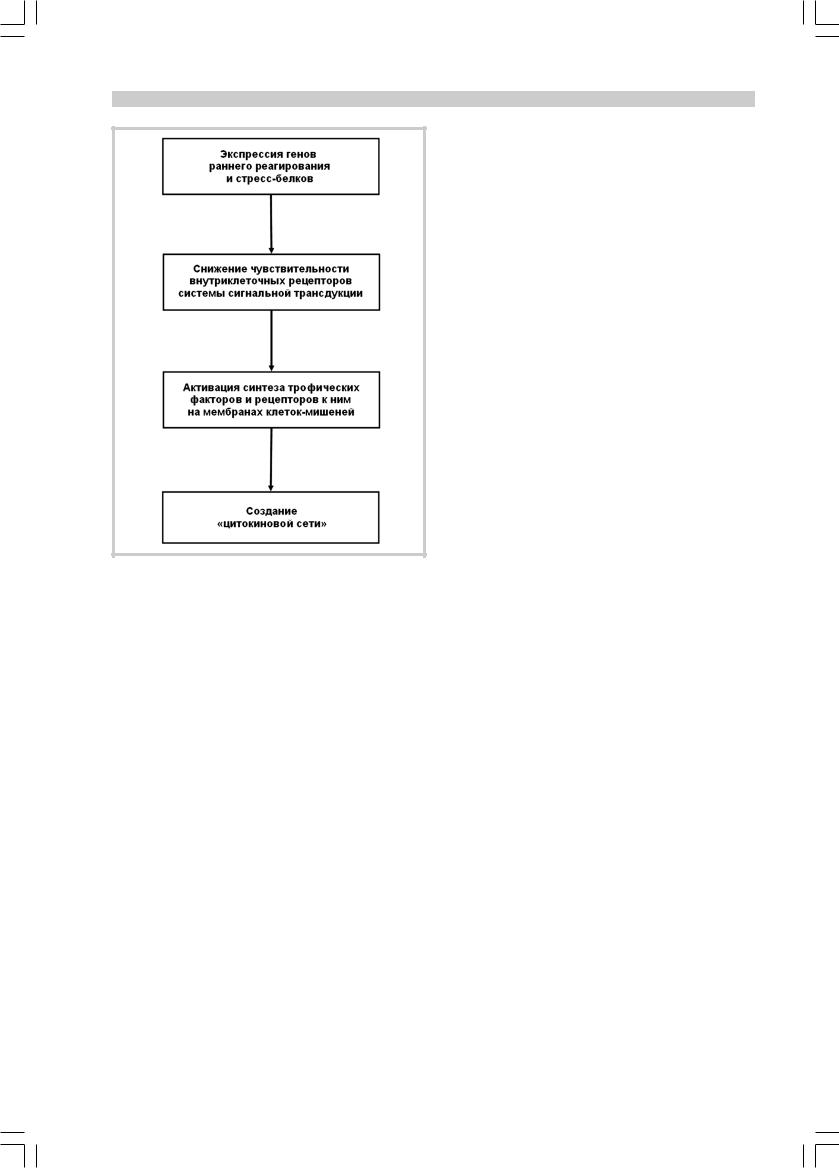
Рис. 5. Схема второго уровня защиты клетки от ишемического повреждения.
Экспозиция кортикальных нейронов с небольшими концентрациями ионизированного кальция индуцировала морфологическое повреждение преимущественно аксонов и запускала последующий апоптоз [45]. При экспозиции с эксайтотоксинами нейроны противостояли глутаматному повреждению и в последующем подвергались апоптозу в связи с тем, что лишались доставки трофических факторов по синапсам [27].
Промежуточные метаболиты системы оксидантного стресса, являющиеся вторичными мессенджерами, инициируют каскад жизнеспасающих реакций в ишемизированной ткани [57]. Их фосфорилирование служит сигналом к экспрессии генов раннего реагирования в пораженных нейронах и глиальных клетках, которая происходит даже в условиях угнетенного (вследствие нарушений чрезмембранного транспорта аминокислот и разрушения рибосом) синтеза протеинов. Экспрессия генов раннего реагирования вызывает образование факторов транскрипции, таких как индуктор фактора роста нервов А (NGFI-A), белки fos, jun, c-myc генных семейств, транскрипционный фактор рецептора стероидного гормона NGFI-B. Факторы транскрипции могут играть важную роль в продукции острофазных стрессбелков. Так, экспрессия «цинк-пальцевых» генов приводит к синтезу стресс-белка Hsp72, предположительно обладающего нейропротективными свойствами. Ферменты супероксиддисмутаза, каталаза, орнитиндекарбоксилаза в настоящее время также отнесены к стресс-белкам, хотя их действительная роль еще до конца не ясна [57].
Экспрессия генов раннего реагирования, индукция образования факторов транскрипции и стресс-белков могут повышать резистентность пораженных клеток мозга к вызванным артериальной гипертонией ишемическим изменениям. Основополагающим механизмом, лежащим в основе
АРТЕРИАЛЬНАЯ ГИПЕРТОНИЯ И ГОЛОВНОЙ МОЗГ
приобретения клеткой толерантности к вторичной ишемии, является снижение чувствительности внутриклеточных рецепторов системы сигнальной трансдукции [44]. Однако если этот организованный ответ не позволяет клеткам мозга избежать предстоящей смерти, включается второй уровень защиты — активируется синтез разнообразных трофических факторов и рецепторов к ним на поверхности мембран кле- ток-мишеней (рис. 5).
Хроническая гипертоническая энцефалопатия
Длительная артериальная гипертония приводит к диффузным патологическим изменениям ткани головного мозга, развивающимся по общим механизмам «нейродегенеративного» процесса с формированием хронической гипертензивной энцефалопатии (по МКБ-10, рубрика I67.4). В зависимости от превалирования того или иного молекулярного механизма могут развиваться разные по морфологии и клиническим проявлениям патологические процессы.
При преобладании нарушений микроциркуляторно-кле- точных взаимодействий и стойких нарушений гемореологии в глубине белого вещества головного мозга формируются мелкие лакуны (до 12 мм в диаметре), являющиеся результатом артериолярных и мелких артериальных инфарктов (лакунарных ишемических инсультов). Глубинная локализация лакун связана с особенностями ангиоархитектоники головного мозга.
Доминирование воспалительно-аутоиммунного компонента приводит к диффузному поражению перивентрикулярного белого вещества головного мозга с характерными признаками на МРТ головного мозга в виде зон лейкоарейоза, преимущественно лобной локализации. Такое повреждение влечет за собой функциональный разрыв префронтальных субкортикальных связей, играющих существенную роль для осуществления психомоторных функций [82]. Недавно показано, что изменения в таламусе, преимущественно в его вентромедиальных ядрах, имеют наибольшую значимость в развитии у больных когнитивных расстройств [25].
Типичными клиническими проявлениями хронической недостаточности мозгового кровообращения на фоне артериальной гипертонии являются жалобы на головокружение, шум в голове, головную боль, снижение памяти. Больным трудно переключаться с одного вида деятельности на другой. Они становятся раздражительны, слезливы. Настроение у них часто подавленное. Наряду с субъективными жалобами выявляются микроочаговые симптомы поражения головного мозга: легкая дизартрия, патологические рефлексы, брадикинезия. При обширном поражении развиваются псевдобульбарный, мозжечковый или пирамидный синдромы, нередко возникают пароксизмальные состояния (падения, обмороки, эпилептические припадки). Длительная артериальная гипертония может приводить к значительному ухудшению когнитивных функций и развитию деменции [3].
В настоящее время принято выделять два наиболее часто встречаемых состояния сосудистой этиологии, сопровождающихся когнитивным дефицитом: деменция сосудистого типа и болезнь Бинсвангера. Эти состояния имеют сходную клиническую картину и часто выявляются у одного и того же больного. Однако учитывая строгую связь деменции сосудистого типа и болезни Бинсвангера с поражением очер- ченных сосудистых бассейнов и характерной патоморфологической картиной, в последние годы была обоснована их нозологическая самостоятельность [3].
Болезнь Бинсвангера (прогрессирующая сосудистая лейкоэнцефалопатия, по МКБ-10, рубрика I67.3) представляет собой особую форму хронического прогрессирующего сосудистого заболевания головного мозга, основным клиническим проявлением которого служит деменция в соче- тании с разнообразной неврологической симптоматикой (подкорковый, мозжечковый, псевдобульбарный и пирамидный синдромы, нарушение функции тазовых органов и др.).
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 10, 2006 |
75 |

ОБЗОРЫ
В основе заболевания и в первую очередь синдрома деменции лежит диффузное поражение белого вещества полушария мозга, которое, по данным нейровизуализирующих методов исследования, выглядит как двустороннее пятнистое или диффузное снижение плотности белого вещества на компьютерных томограммах или гиперинтенсивность сигнала на магнитно-резонансных томограммах в Т2-режиме (лейкоарейоз) [2].
Сосудистая деменция (по МКБ-10, F01), называемая также «мультиинфарктной» вследствие основного морфологического признака — наличия множественных мелких инфарктов в белом веществе головного мозга, клинически проявляется ухудшением в интеллектуальной сфере (снижением памяти, замедлением мышления, дефицитом внимания), расстройствами вегетативной регуляции (головокружения, головные боли, бессонница), а также эмоциональными изменениями (колебания настроения, отсутствие заинтересованности, проявления гневливости). В отли- чие от болезни Бинсвангера течение заболевания характеризуется не постепенным, а «ступенеобразным» нарастанием симптомов, при этом в основе каждого ухудшения лежит развитие нового лакунарного инфаркта головного мозга.
Результаты популяционных исследований подтвердили участие артериальной гипертонии в формировании когнитивного дефицита и деменций разного типа [29], которые в настоящее время оказывают значимое негативное влияние
на социальную и экономическую сферы общественной жизни [39]. Так, в крупном многоцентровом исследовании SCOPE, посвященном вопросам возникновения и прогрессирования деменции и включавшем 1810 пациентов, было показано, что больные с АГ, получавшие гипотензивные препараты, достоверно реже страдали когнитивными расстройствами, чем пациенты, которым коррекция АД не проводилась [65]. По данным испытания SYSTEUR, на фоне применения антагонистов кальция в комбинации с ингибиторами ангиотензинпревращающего фермента (АПФ) в 50% случаев удалось предотвратить развитие деменции [42, 59]. Исследование PROGRESS, включавшее 6105 больных с перенесенными ранее нарушениями мозгового кровообращения, показало, что активная гипотензивная терапия периндоприлом в сочетании (или без) с индопамидом значи- мо снижает риск развития постинсультной деменции и когнитивных нарушений [31]. В то же время результаты исследования SHEP, в котором больным проводили антигипертензивную терапию диуретиками, не обнаружили эффективности снижения АД для предотвращения нарушения когнитивных функций [104], что подчеркивает значимость ренин-ангиотензиновой системы во вредоносном действии артериальной гипертонии на головной мозг.
Доказанная связь между уровнем АД и наличием деменции в позднем возрасте предполагает рассматривать АГ как поддающийся коррекции фактор для предотвращения «беспомощной старости» [16, 18].
ЛИТЕРАТУРА
1.Березин В.А. Нейрохимия 1990; 1: 1: 114—123.
2.Верещагин Н.В., Калашникова Л.А., Гулевская Т.С., Миловидов Ю.К.
Журн неврол и психиат 1995; 1: 98—103.
3.Гринштейн А.Б., Шнайдер Н.А. Цереброваскулярные осложнения артериальной гипертонии. Красноярск: КГМА 2002; 160.
4.Гусев Е.И., Скворцова В.И. Ишемия головного мозга. М: Медицина 2001; 327.
5.Гусев Е.И., Скворцова В.И., Изыкенова Г.А. и др. Изучение уровня аутоантител к глутаматным рецепторам в сыворотке крови у больных в остром периоде ишемического инсульта. Журн неврол и психиат 1996; 5: 68—72.
6.Кушаковский М.С. Гипертоническая болезнь (эссенциальная гипертония): причины, механизмы, клиника, лечение. Ст-Пе- тербург: СОТИС 1995; 320.
7.Малашхия Ю.А. Иммунология 1990; 3: 12—15.
хронической ишемией головного мозга. В кн.: Современные подходы к диагностике и лечению нервных и психических заболеваний. Ст-Петербург 2000; 341.
18.Чазова И.Е. Инсульт. (Приложение). Журн неврол и психиат 2001; 3: 3—7.
19.Чехонин В.П., Рябухин И.А., Белопасов В.В. и др. Моноклональные антитела в нейробиологии. Новосибирск 1995; 160—170.
20.Шевченко О.П. и др. Артериальная гипертония и церебральный инсульт. М: Реафарм 2001; 187.
21.Шерстнев В.В. Дис. ... …ä-ðà ìåä. íàóê. Ì 1983.
22.Штарк М.Б. Мозгоспецифические белки (антигены) и функция нейрона. М 1989.
23.Akins P.Ò., Liu P.K., Hsu C.Y. Immediate Early Gene Expression in Response to Cerebral Ischemia: Friend or Foe? Stroke 1996; 27: 1682—1687.
8.Малашхия Ю.А. Интерн журн иммунол реабилитол 1995; 1: 10— 24. Arvin Â., Neville L.F., Barone F.C., Feuerstein G.Z. Neurosci Biobe-
17.
9.Писаренко О.Н., Студнева И.М., Постнов А.Ю. Особенности энергетического состояния тканей при спонтанной гипертензии крыс (SHR). Кардиология 1998; 12: 37—40.
10.Полетаев А.Б. Дис. ... …ä-ðà ìåä. íàóê. Ì 1984.
11.Полетаев А.Б., Шерстнев В.В. Успехи современной биологии 1987; 103: 1: 124—132.
12.Постнов Ю.В. К истокам первичной гипертонии: подход с позиции биоэнергетики. Кардиология 1998; 12: 41—48.
13.Скворцова В.И. Участие апоптоза в формировании инфаркта мозга. Инсульт. (Приложение). Журн неврол и психиат 2001; 2: 12—19.
14.Скворцова В.И., Раевский К.С., Коваленко А.В. и др. Содержание нейротрансмиттерных аминокислот в спинномозговой жидкости больных острым ишемическим инсультом. Журн неврол и психиат 1999; 2: 34—39.
15.Скворцова В.И., Насонов У.Л., Журавлева Е.Ю. и др. Клиникоиммунобиохимический мониторинг факторов локального воспаления в остром периоде полушарного ишемического инсульта. Журн неврол и психиат 1999; 5: 27—32.
16.Скворцова В.И., Чазова И.Е., Стаховская Л.В. Вторичная профилактика инсульта. М: ПАГРИ 2002; 120.
17.Хаджиева М.Х., Скворцова В.И., Шерстнев В.В. и др. Изучение нейротрофических факторов и аутоантител к ним у больных с
hav Rev 1996; 20: 3: 445—452.
25.Baezner H. Daffertshofer M. Subcortical vascular encephalopathy. Ther Umschr 2003; 60: 9: 541—552.
26.Banati R.B., Gehrmann J., Kreutlberg G.W. In: Cellular and molecular mechanisms of ischemic brain damage (K. Siesjo, T. Wieloch, eds.). 1996; 329—337.
27.Bindokas V.P., Miller R.J. Excitotoxic degeneration in initiated at non-random sites in cultured rat cerebellar neurons. J Neurosci 1995;
15:6999—7011.
28.Blinzinger K., Kreutzberg G. Displacement of synaptic terminals from regenerating motoneurons by microglial cells. Z Zellforsch Mikrosk Anat 1968; 85: 145—157.
29.Bodenmann P., Ghika J., van Melle G., Bogousslavsky J. Neurological comorbidity in parkinsonism. Rev Neurol 2001; 187: 1: 45.
30.Carlos T.M., Schwartz B.R., Kovach N.L. et al. Vascular cell adhesion molecule-1 mediates lymphocyte adherence to cytokine-activated cultured human endothelial cells. Blood 1990; 76: 5: 965—970.
31.Chalmers J., MacMahon S. Perindopril pROtection aGains REcureent Stroke Stusy (PROGRESS): interpretation and implementation. J Hypertens 2003; 21: 5: Suppl: 9—14.
32.Chen L., Tian X., Song L. Chin Med J (Engl) 1995; 108: 5: 361—366.
33.Chowdhary S., Townend J.N. Nitric oxide and hypertension: not just an endothelium derived relaxing factor! J Hum Hypertens 2001; 15:
4:219—227.
76 |
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 10, 2006 |

АРТЕРИАЛЬНАЯ ГИПЕРТОНИЯ И ГОЛОВНОЙ МОЗГ
34.Clark R.S., Kochanek P.M., Schwan M.A. et al. Inducible nitric oxide synthase expression in cerebrovascular smooth muscle and neutrophils after traumatic brain injury in immature rats. Pediat Res 1996; 39: 5: 784—790.
60.Kuang F., Wang B.R., Zhang P. et al. Extravasation of blood-borne immunoglobulin G through blood-brain barrier during adrenalineinduced transient hypertension in the rat. Int J Neurosci 2004; 114: 6: 575—591.
35.Cominacini L., Pasini A., Garbin U. et al. Zofenopril inhibits the ex61. Lassmann H., Zimprich F., Rossler K., Vass K. Inflammation in the
pression of adhesion molecules on endothelial cells by reducing reactive oxygen species. Am J Hypertens 2002; 15: 10: 1: 891—895.
36.De Vries H.E.,Kuiper J.,De Boer A.G. et al. The blood-brain barrier in neuroinflammatory diseases. Pharmacol Rev 1997; 49: 2: 143— 155.
37.De Vries H.E., Moor A.C., Blom-Roosemalen M.C. et al. Lymphocyte adhesion to brain capillary endothelial cells in vitro. J Neuroimmunol 1994; 52: 1: 1—8.
38.Ericsson A., Liu C., Hart R.P., Sawchenko P.E. Type 1 interleukin-1 receptor in the rat brain: distribution, regulation, and relationship to sites of IL-1-induced cellular activation. J Comp Neuro 1995;
361:681—698.
39.Erkinjuntti T. Subcortical vascular dementia. Cerebrovasc Dis 2002;
13:2: 58—60.
40.Fabry Z., Waldschmidt M.M., Hendrickson D. et al. Adhesion molecules on murine brain microvascular endothelial cells: expression and regulation of ICAM-1 and Lgp 55. J Neuroimmunol 1992; 36:
1:1—11.
41.Festoff B.W., Israel R.S., Engel W.K. J Neurol 1977; 27: 963—970.
42.Frishman W.H. Heart Dis 2002; 4: 6: 380—386.
43.Giulian D. Reactive glia as rivals in regulating neuronal survival. Glia 1993; 7: 1: 102—110.
44.Gusev E.I., Skvortsova V.I. Brain Ischemia 2003. New York—Lon- don—Berlin: Kluwer Academic Publishers 382.
45.Gwag B.J., Canzoniero L.M., Sensi S.L. Calcium ionophores can induce either apoptosis or necrosis in cultured cortical neurons. Neurosci 1999; 90: 4: 1339—1348.
46.Hawkins C.P., Munro P.M., MacKenzie F. et al. Duration and selectivity of blood-brain barrier breakdown in chronic relapsing experimental allergic encephalomyelitis studied by gadolinium-DTPA and protein markers. Brain 1990; 113: 365—378.
47.Hirooka Y., Head G.A., Potts P.D. et al. Medullarey neurons activated by angiotensin II in the conscious rabbit. Hypertension 1996; 27: 287—296.
48.Hohfield A. Ann Neurol 1989; 25: 531—538.
49.Iadecola C., Zhang P., Casey R. et al. Inducible nitric oxide synthase gene expression in vascular cells after transient focal cerebral ischemia. Stroke 1996; 27: 1373—1380.
50.Ito H., Takemori K., Kawai J., Suzuki T. AT1 receptor antagonist prevents brain edema without lowering blood pressure. Acta Neurochir 2000; 76: Suppl: 141—145.
51.Ito K., Hirooka Y., Sakai K. et al. Rho/Rho-kinase pathqay in brain stem contributes to bllod pressure regulation via sympathetic nervous system: possible involvement in neural mechanisms of hypertension. Circ Res 2003; 92: 1337—1343.
52.Kanoh Y., Ohtani H. Levels of interleukin-6, CRP and alpha 2 macroglobulin in cerebrospinal fluid (CSF) and serum as indicator of blood-CSF barrier damage. Biochem Mol Biol Int 1997; 43: 2: 269— 278.
53.Klahr S., Morrissey J. Amgiotensiin II an gene expression in the kidney. Am J Kidney Dis 1998; 31: 1: 171—176.
54.Kataoka C., Egashira K., Inoue S. et al. Important role of Rho-kinase in the pathogenesis of cardiovascular inflammation and remodeling induced by long-term blockade of nitric oxide sunthesis in rats. Hypertension 2002; 39: 245—250.
55.Kiarash A., Pagano P.J., Tayeh M. et al. Upregulated Expression of Rat Heart Intercellular Adhesion Molecule-q in Angiotensin IIbut Not PhenylephrineInduced Hepartension. Hypertension 2001;
37:1: 58—65.
nervous system. Basic mechanisms and immunological conepts. Rev Neurol (Paris) 1991; 147: 12: 763—781.
62.Lee S.J., Drabik K., Van Wagoner N.J. et al. ICAM-1-induced expression of proinflammatory cytokines in astrocytes: involvement of extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways. J Immunol 2000; 165: 8: 4658—4666.
63.Linington C., Bradl M., Lassmann H. et al. Augmentation of demyelination in rat acute allergic encephalomyelitis by circulationg mouse monoclonal antibodies directed against a myelin/oligodendrocyte glycoprotein. Am J Pathol 1988; 130: 3: 443—454.
64.Linnik M.D., Zahos P., Geschwind M.D., Federoff H.J. Expression of bcl-2 from a defective herpes simplex virus-1 vector limits neuromal death in focal cerebral ischemia. Stroke 1995; 26: 1670—1675.
65.Lithell H., Hansson L., Skoog I. The Study on COgnition and Peognosis in the Elderly (SCOPE); outcomes in patients not receiving add-on therapy after randomization. J Hypertens 2004; 22: 8: 1605— 1612.
66.Liu Ò., Dark R. K., McDonnell P.C. et al. Tumor necrosis factor-alpha expression in ischemic neurons. Stroke 1994; 25: 1481—1488.
67.MacManus J.P., Linnik M.D. Gene expression induced by cerebral ischemia: an apoptotic perspective. J Cereb Blood Flow Metab 1997; 17: 815—832.
68.Male D., Pryce G., Linke A., Rahman J. Lymphocyte migration into the CNS modeled in vitro. J Neuroimmunol 1992; 40: 167—172.
69.Matsumoto K., Graf R. et al. Elevation of neuroactive substances in the cortex of cats during prolonged focal ischemia. J Cerebr Blood Flow Metab 1993; 13: 586—594.
70.McGeer P.L., Kawamala Ò., Walker D.G. et al. Glia 1993; 7: 84—92.
71.Michaels R.L., Rothman S.M. Glutamate neurotoxicity in vitro: antagonist pharmacology and intracellular calcium concentrati. J Neurosci 1990; 10: 283—292.
72.Mohacsi A., Magyar J., Tamas B., Nanasi P.P. Effects of endothelins un csrdiac and vascular cells: new therapeutic target for the future? Curr Vasc Pharmacol 2004; 2: 1: 53—63.
73.Molkentin J.D., Lu J.-R., Anros C.L. et al. Cell 1988; 93: 215—228.
74.Morgan J.I.,Curran T. Stimulus-transcription coupling in the nervous sustem: involvement of the inducible proto-oncogenes fos and jun. Ann Rev Neurol 1991; 14: 421—451.
75.Muller D.N., Dechend R., Mervaala E.M. et al. NF-kappaB inhibition ameliorates angiotensin II-induced inflammatory damage in rats. Hypertension 2000; 35: 1: 2: 193—201.
76.Muller D.N., Mervaala E.M., Schmidt F. et al. Effect of bosentan on NF-kappsB, inflammation, and tissue factor in angiotensin II-in- duced end-organ damage. Hypertension 2000; 36: 2: 282—290.
77.Murota S., Fujita H., Morita I. Involvement of adhesion molecules in vascular endothelial cell injury by oxygen radicals released from activated leukocytes. In: Intractable vasculitis syndromes (T. Tanabe, eds.). Hokkaido University Press 1993; 115—122.
78.Nawroth P.P., Stem D.M. Exp Med J 1986; 163: 740—745.
79.Nogawa S., Zhang F., Ross M.E., Iadecola C. Cyclo-oxygenase-2 gene expression in neurons contributes to ischemic brain damage. J Neurosci 1997; 17: 2746—2755.
80.Nordan R., Potter M. A macrophage-derived factor required by plasmacutomas for survival and proliferation in vitro. Science 1986; 233: 4763: 566—569.
81.Nowak T.S., Kiessling Jr. et al. M. Reprogramming of Gene Expression after Ischemia. In: Cerebral Ischemia (Walz Wolfgang, ed.). New Jersey, Totowa, Humana Press 1999; 145—217.
56.Kim J.S. J Neurol Sci 1996; 137: 69—78.
57.Kogure Ê., Yamasaki Y., Matsuo Y. et al. Inflammation of brain after ischemia. Acta Neurochir 1996; 66: 40—43.
58.Kojima K., Berger T., Lassmann H. et al. Experimental autoimmune panencephalitis and uveoretinitis transferred to the Lewis rat by T lymphocytes specific for the S100 beta molecule, a calcium binding protein of astroglia. J Exp Med 1994; 180: 3: 817—829.
59.Kossmann Ò., Morganti-Kossmann Ñ., Trent. Î. et al. Shock 1995; 4: 5: 311—317.
82.Oggunniyi A., Talabi O. Cerebrovascular complications of hypertension. Niger J Med 2001; 10: 4: 158—161.
83.Oppenheim R.W. Cell death during development of the nervous system. Ann Rev Neurosci 1991; 14: 453—501.
84.Pechanova O., Dobesova Z., Cejka J. et al. Vasoactive systems in L- NAME hypertension: the role of inducible metric oxide synthase. J Hypertens 2004; 22: 1: 167—173.
85.Petito C.K., Morgello S., Felix J., Lesser M.L. J Cereb Blood Flow Metab 1990; 10: 850—859.
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 10, 2006 |
77 |