


Signal Transduction
Development of imatinib, inhibitor of c-Abl
Imatinib (Gleevec) emerged from a random screen of 2- phenylaminopyrimidine compounds, known to be inhibitors of PKC- , as possible inhibitors of PDGF-R tyrosine kinase. The aim was to discover
compounds that could suppress the growth of PDGF-R-activated cell lines. The test also included the oncogenic v-Abl as a counter screen, because of its very distant sequence resemblance to PDGF-R. It came as a surprise that the most potent inhibitor of PDGF-R kinase activity, CGP 531716, was also a potent inhibitor of v-Abl. With new potential treatments in mind, it was
further optimized for v-Abl kinase inhibition, the compound CGP 57148B, now known as imatinib or by its trade names Gleevec and Glivec, emerging as the most effective (Figure 23.8) (see Druker and Lydon40 for review). Importantly, imatinib has selective and potent activity in cells possessing the Philadelphia chromosome and yet spares normal cells.41,42
Imatinib was first tested in patients who were in the chronic phase of CML and refractory to IFNtherapy. In one trial, complete normalization of the cellular composition of the blood was obtained after 4 weeks of treatment in 53 out of 54 patients treated in the higher dose range (the drug is seemingly benign: a maximal tolerated dose was not identified and adverse effects were minimal).43 The amount of detectable Bcr-Abl fused genes in bone marrow cells was considerably reduced and the patients remained free of disease
Despite tight rules and regulations, the reasons why a particular drug might or might not gain approval and why, once approved, it might still be unsuccessful, depends on several factors that are hard to control.
It is possible that adverse effects are not reported to the approval authorities, or that their importance may not be recognized. They may even go unrecognized. Adverse effects may eventually come to light only after prolonged and largescale application. Drugs may, once firmly established, also be prescribed to patients for whom they are not indicated. In a number of cases they may have unexpected adverse effects or lack therapeutic advantage because patients
do not follow the dosage protocols or take over-the-counter, non-prescribed remedies alongside.
We have heard it said that in England during the 1990s, patients spent more on non-prescription remedies than the entire budget of the Medical Research Council. With a rapidly growing and uncontrolled Internet market this can only
get worse. Some drugs that have only marginal therapeutic effects gain approval because of the immense pressure applied not only by the pharmaceutical companies to sell their products, but also by patients, newspapers, lawyers, and politicians who demand instant treatment of life-threatening illnesses.
Finally, reasonable or unreasonable litigation may damage the reputation of a company to the extent that drugs have to be taken off the market: the Vioxx drama is a case in point (see http://www.vioxx-recall-lawsuit.com/).
750
Targeting Transduction Pathways for Research and Medical Intervention
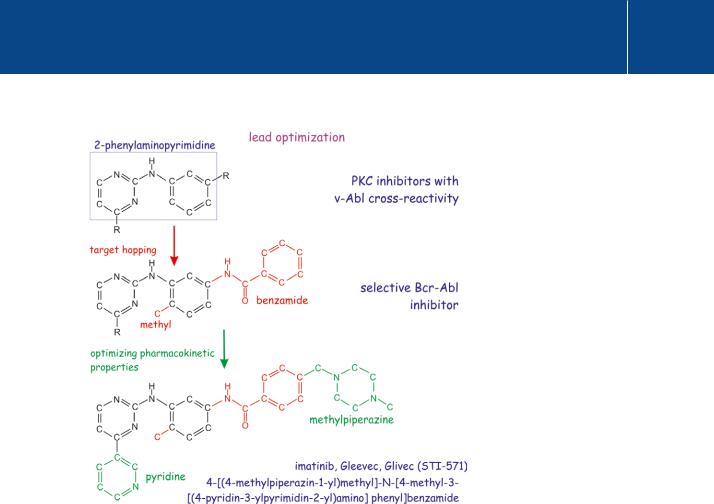
Fig 23.8 Optimization of imatinib as a chemotherapeutic agent.
The discovery that 2-phenylaminopyrimidine inhibitors of PKC also inhibit the unrelated v-Abl oncogene turned attention to its potential use in the treatment of chronic myelogenous leukaemia. Starting with the 2-phenylaminopyrimidine backbone, addition of the benzamidine group increased activity against tyrosine kinases, the methyl group reduced its activity against PKC (so-called ‘target hopping’). Addition of a 3 -pyridyl group improved the activity in cellular assays. Subsequent addition of N-methylpiperazine increased water solubility and oral bioavailability, enabling the drug to survive the stomach and to enter the bloodstream.
for over 1 year. Remarkable results were also obtained in subsequent largerscale trials. Most, 85%, showed a complete cytogenetic response. In assays employing in situ hybridization with fluorescent probes, the Bcr-Abl fusion protein became undetectable and the Philadelphia chromosome was absent.
However, even with all this, it does not follow that the tumour cells were completely eliminated, as Bcr-Abl transcripts remained detectable by reverse transcriptase PCR in 96% of the responding patients. There remains a potential pool of cells from which resistant clones can emerge at a later date. Indeed, after 42 months of follow-up, relapse had occurred in 16% of these patients.44 The response is much less promising in patients who have reached blast crisis.43 In some cases this resistance to treatment is due to a very high
751

Signal Transduction
c-Abl is not the sole target of imatinib.
Good activity is also found against PDGF-R , c-KIT, Lck, C-Raf, and the three VEGF receptors. Because of its strong inhibitory action on c-KIT (a tyrosine kinase receptor), it is
also approved for the treatment of inoperable KIT-positive and metastatic gastrointestinal stromal tumours (GIST). Although it is generally well tolerated, there are some cases in which prolonged treatment causes congestive heart failure. This is perhaps a more general adverse effect of tyrosine kinase inhibitors which seem
to cause cardiomyocyte dysfuntion and/or cell death.47
level of Bcr-Abl expression, in others to mutations of Bcr-Abl.45,46 With the aim of overcoming drug resistance, new ATP competitors, specific for Bcr-Abl, are currently under development.
Why is treatment of CML successful?
In comparison with other drugs that target EGF receptors, the success of imatinib is not easy to understand, but several arguments can be advanced to explain why targeting Bcr-Abl in CML has been fruitful. The first, despite a long list of reasons predicting that it would work, is simply pure luck. A second
argument may be that blood-borne cells differ fundamentally from solid tissue cells. These can switch from being attached to free floating without undergoing the risk of apoptosis, and from this we understand that they have alternative survival pathways (see page 400). Because of their enhanced survival capacity, fewer hits may be required in order to create a leukaemia, but on the other hand, this may have the advantage that there will also be fewer disease genes to target for therapy. A third argument is that CML generally comes to notice in its early chronic benign phase, at a point when many solid tissue tumours
would be undetectable. Moreover, because of their very high proliferation rates, somatic mutations in stem cells may be (at least in their initial phase) more likely to give rise to‘monoclonal’tumours. Polyclonality is certainly one of the hurdles that must be overcome in the treatment of solid tissue tumours.
Molecular mechanism of inhibition by imatinib
Imatinib binds in the catalytic cleft of c-Abl, acting as a competitive inhibitor of ATP binding (Figure 23.6). It binds most efficiently to partially activated c-Abl, in which the SH2/SH3 clamp is removed, but the tyrosine (Y412) in the activation segment is not phosphorylated. The drug seeks out most effectively those malignant cells that depend acutely on sustained Bcr-Abl kinase activity for their survival. By precipitating their apoptosis, imatinib greatly reduces the disease burden (1000-fold reduction in cells expressing Bcr-Abl). However, as indicated above, a residue of disease cells remains and some patients never respond at all. This is because leukaemic stem cells, being either quiescent or protected by the bone marrow micro-environment, escape the drug. Alternatively, the resistance may be due to mutations in Bcr-Abl or Bcr-Abl amplification.
Resistance due to mutations at different sites in the Abl moiety of Bcr-Abl
Analysis of resistant cells has revealed a whole range of mutations (Figure 23.9). Some of these act at a distance from the catalytic site and have an allosteric influence on drug binding. They destabilize the autoinhibited conformation of the Abl kinase to which the drug binds, and shift the equilibrium toward the active kinase conformation. This precludes drug binding, but not ATP binding. Other mutations directly involve drug binding. These are P-loop mutations (Y253 or E255), activation segment mutations or mutations that hinder drug occupancy (T315).
752

Targeting Transduction Pathways for Research and Medical Intervention
Fig 23.9 Mutations in Bcr-Abl that cause insensitivity to imatinib (drug resistance).
Numerous mutations in the Abl moiety have been identified in patients resistant to treatment with imatinib. Only a few of these are direct contact residues; others are remote from the catalytic cleft and their effect is not easily explained. In a survey of 177 patients having such resistance, 28 mutations were identified, 4 of which were predominant (T315I (37), E255K (34), M351I (27), Y253H (12)). T315, the ‘gatekeeper’ of imatinib, is a key contact residue in the binding pocket. Y253 and E255 regulate the positioning of the P-loop, which acts as a lid on the drug-binding site. How M351 causes resistance is not known. (1opj37).
Mutant information from Deininger et al.48
More (many more, about 112 resistance mutations in total) were revealed in an ingenious experiment in which the Bcr-Abl gene was randomly
mutagenized by insertion into a highly labile strain of E. coli. The altered genes were then screened for resistance in BaF3 cells which are normally dependent on the presence of IL-3 for their survival.49 Cells expressing deregulated forms of Bcr-Abl could grow in the absence of IL3 and of these, some could grow in the presence of imatinib.50 The mutants identified in this way overlap all the mutants detected in non-responding patients or in patients suffering relapse. We conclude that tyrosine protein kinases are highly plastic entities that can adopt many inactive states so long as they fulfil certain precise structural criteria as they converge towards their active conformation.
A similar conclusion had already been drawn from structural studies of serine/threonine kinases such as MAPkinase, CDK2, and PKA. Their structural conformations differ considerably in their inactive states and also differ with respect to their activation mechanisms. However, their active conformations display clear similarities (for review see Johnson and Lewis.51)
All this provides a challenge for drug development because those drugs that act on protein kinases in their inactive conformation, while necessarily specific, may more easily suffer from resistance mutations. By contrast, drugs
binding to active conformations are likely to be less specific, but may offer the
753