

Books for lectures / Gompert Signal Transd / Ch19 PKC revisited
.pdfChapter 19
Protein Kinase C Revisited
PKC in cell transformation
Long before their association with PKC became apparent, it was evident that extracts of croton oil can act as cocarcinogens, enhancing the tumorigenic activity of other substances such as benzo[a]pyrene.1 In some
culture systems the phorbol ester PMA increases the number of mitoses,2 but in the absence of a tumour promoter it fails to effect full cell transformation. One might think that PKC should be implicated in all this, maybe acting to phosphorylate a transcription factor to induce specific gene expression. However, the matter remains unresolved and PKC isoforms responsive to phorbol ester have failed to qualify as true oncogenes.
Despite a voluminous literature, understanding of the relationship between PKC and tumour formation remains somewhat hazy. At best, it seems to have accessory roles, boosting the activity of the real transformers such as Ras and Raf. Since more and more information focuses on the action of atypical
The five-ring polycyclic aromatic hydrocarbon benzo[a]pyrene is highly carcinogenic. It is the component of coal tar responsible for the cancers (sooty warts, cancers of the scrotum) suffered by chimney sweeps in 18th century England.3 To this day
it presents a health hazard as a carcinogen present in cigarette and marijuana smoke, and possibly in barbecued
577

Signal Transduction
meat. As with many other chemical carcinogens, benzopyrene is
better regarded as a procarcinogen,
requiring conversion to an active derivative, benzo[a]pyrene diol epoxide. This slots into the major groove of DNA,4 specifically
targeting the p53 gene.5
The many abbreviations used in this chapter are collected together at the end of the chapter.
PKC( , ) in the regulation of cell polarity, we concentrate on this aspect of PKC signalling.
With the aim of understanding the mechanisms underlying tumour promotion by phorbol esters, two independent strategies have been applied. By searching for transcriptional control elements that mediate the phorbol-ester-induced alterations in gene expression, it should be possible to work backwards, identifying first the transcription factor(s) that bind these elements and then the signal transduction pathway that regulates their activation.6 The alternative approach has been to over-express various isoforms of PKC and to study changes in cell phenotype.7
The search for transcription factors that mediate phorbol ester effects
From TRE to SRE
Analysis of the promoter regions of a number of genes (for instance, collagenase, metallothionein IIA, and stromelysin) induced by phorbol ester reveals a conserved 7 bp palindromic motif, the TPA-responsive element (TRE). This is recognized by AP-1, understood to be at the receiving end of a complex pathway that transmits the effects of phorbol ester tumour promoters from the plasma membrane to the transcriptional machinery, possibly involving protein kinase C.8 AP-1 collectively describes a group of structurally and functionally related members of the Jun and Fos protein family, together
with some members of the ATF and MAF families (in combination with Jun proteins)9 and linked by a leucine zipper protein–protein interaction motif (see Figure 19.1 and Tables 19.1 and 19.2).10 These transcription factors are similar in structure to CREB (see page 248).11–14 CREB can also bind to the TRE13 and so inhibit activation through c-Jun.
AP-1
The activator protein-1 transcription factor complexes owed their initial definition to a fortuitous convergence of tumour virology and transcription factor research. AP-1 had been described as a TPA-inducible (or PMAinducible) activity that addresses specific sequences in the enhancer of the metallothionein gene and in the 72 bp repeat of the simian virus 40 enhancerregion. The oncoproteins v-Fos and v-Jun were identified as transforming proteins in the Finkel–Biskis–Jinkins murine osteosarcoma virus and in avian sarcoma virus-17, respectively. v-Jun was linked with AP-1 because of its homology with the yeast transcriptional regulator GCN4, which binds an identical DNA sequence. Indeed, the DNA-binding domains of Jun and of GCN4 are exchangeable. The cellular homologue of v-Fos-associates with a protein of 39 kDa, recognized as c-Jun. This finding identified Fos and Jun as partners, together forming the AP-1 complex that binds DNA.15
578
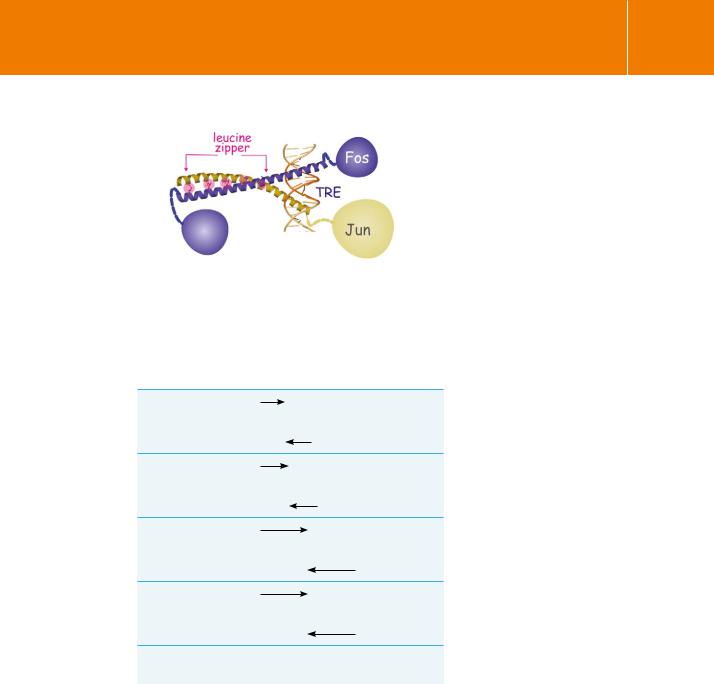
Protein Kinase C Revisited
Fig 19.1 Activator protein-1 (AP-1) complexes.
Jun and Fos, members of AP-1 complex proteins, are connected through numerous leucine residues, the leucine zipper. Here, they bind DNA at the palindromic -TGACTACA-/-ACTGAGT- sequence (TRE)Image adapted from Eferl and Wagner.16
Table 19.1 Examples of palindromic DNA sequences recognized by AP1 factors
TRE
-TGACTCA-
-ACTGAGT-
CRE
-TGACGTCA-
-ACTGCAGT-
MAREI
-TCGTCACGTCAGCA-
-ACGACTGCAGTCGT-
MAREII
-TGCTGACGTCAGCA-
-ACGACTGCAGTCGT-
ARE |
-a/gTGACnnnGC- |
|
-t/cACTGnnnCG- |
|
|
v-Fos and v-Jun are transforming proteins by virtue of their prolonged half-lives, enhanced transcriptional activity and a change in the spectrum of target genes (compared to their cellular counterparts).11,12 It became apparent that cellular AP-1 is composed of different combinations of proteins, always containing either c-Jun or c-Fos, but not necessarily binding to the TRE. Other response elements recognized by AP-1 are CRE, MARE I, MARE II, and ARE. Possible combinations, with their preferred DNA response element, are listed in Table 19.1.
579
Signal Transduction
Table 19.2 Partners of Jun and Fos in AP-1 complexes
Jun |
|
Fos |
|
|
|
|
|
c-Jun |
TRE CRE |
c-Jun |
TRE CRE |
|
|
|
|
JunB |
TRE CRE |
JunB |
TRE CRE |
|
|
|
|
junD |
TRE CRE |
JunC |
TRE CRE |
|
|
|
|
FosB |
TRE CRE |
|
|
|
|
|
|
Fra1 |
TRE CRE |
|
|
|
|
|
|
FRA2 |
TRE CRE |
|
|
|
|
|
|
ATFa |
CRE TRE |
ATFa |
Non-binding |
|
|
|
|
ATF2 |
CRE TRE |
ATF2 |
CRE TRE |
|
|
|
|
ATF4 |
(CRE) |
ATF4 |
CRE |
|
|
|
|
B-ATF |
TRE CRE |
|
|
|
|
|
|
c-MAF |
(MAREI/II) |
c-MAF |
(MAREI/II) |
|
|
MAFA |
(MAREI/II) |
|
|
MAFB |
(MAREI/II) |
|
|
|
|
NRL |
(TRE-related) |
NRL |
(TRE-related) |
|
|
|
|
MAFF/G/K |
(MAREI/II) |
MAFF/G/K |
(MAREI/II) |
|
|
|
|
NRF1 |
(ARE) |
|
|
|
|
|
|
NRF2 |
(ARE) |
NRF2 |
(ARE) |
|
|
|
|
NFIL-6 |
(ARE) |
NFIL6 |
(ARE) |
|
|
|
|
Although they were originally discovered in the context of tumorigenesis, not all forms of AP-1 cause cell transformation and their effect is dependent on context. Roughly speaking, c-Fos, FosB, and c-Jun are considered oncogenes although they have not been found associated with cancer (no mutants). Fra1 and Fra2 are considered weak oncogenes whereas JunB and junD have tumour suppressor qualities.4,5,16
Regulation of the AP-1 complex
The upstream signal transduction pathway that regulates AP-1 activity proved a hard nut to crack. Initially it appeared that activation of PKC causes dephosphorylation of c-Jun, just in the basic region where it binds DNA.17 Phosphorylation of this segment can also be achieved (in the test tube) by
580

Protein Kinase C Revisited
glycogen synthase kinase-3 (GSK3 ) and so it was postulated that PKC stimulates the binding of c-Jun DNA through inhibition of GSK3 , which would result in dephosphorylation of the basic region. Consistent with this is that activation of PKC ( , 1, 2, and ) causes phosphorylation and thus deactivation of GSK3 .17,18 However the interaction between GSK3 and c-Jun needed verification and the phosphatase that strips the phosphates remained enigmatic. That this could not be the whole story became clear when it was found that phosphorylation of four residues at the N-terminus is far more important for c-Jun-mediated transcriptional activity and cell transformation.19,20
The discovery of a Jun N-terminal protein kinase, JNK-1, which phosphorylates c-Jun,21,22 drew attention away from PKC and on to the newly emerging family of MAP kinases and their effects on the serum response element, SRE (see page 342).
PKC activates Raf through phosphorylation of RKIP
If PKC is not the initiator of TREand SRE-mediated gene transcription, it certainly acts to reinforce the input derived from growth factors and stress conditions (including inflammatory factors). PKC activates the Ras-activated kinase c-Raf, an oncogene that cooperates in the transformation of NIH3T3 fibroblasts23 (see page 327). In rat embryo fibroblasts, activation of Raf-1 is also essential for the transforming effect of PKC .24
Since all growth factors induce the generation of diacylglycerol and hence activate PKC, it follows that PKC reinforces the Ras-mediated growth factor signal, either at the level of Ras itself or at the level of Raf-1. Indeed, PKC phosphorylates the inhibitor of c-Raf, RKIP, causing its detachment from c-Raf and so reinstates the Ras–ERK pathway.25,26 In this way PKC contributes to gene transcription through the SRE (Figure 19.2).27,28
Phorbol ester also activates Ras directly, through membrane binding of the guanine nucleotide exchange factor RasGRP. Again, this pathway is reinforced by PKC through phosphorylation and activation of RasGRP. This feedforward mechanism plays an important role in effective B and T cell signalling.
PKC rewires the ERK pathway: role of RACK1 and JIP
A large proportion of human melanomas harbour a highly active Ras–ERK pathway due to gain-of-function mutations in the genes coding for B-Raf and/ or N-Ras (see page 327 onwards). CyclinD, which drives progression of the cell division cycle in G1, is also up-regulated. The transcription factors c-Jun and ATF2, forming an AP-1 complex, are regulators of cyclinD expression. c-Jun must be phosphorylated in order to be transcriptionally active and this could occur through at least two pathways, the one comprising the Ras–Raf–MEK– ERK cascade,29 the other comprising MKK4 or -7 and JNK. Studies with kinase
With the discovery of the Jun kinases, PKC lost any claim or right of ownership over specific pathways related to cell transformation. This is a
point of interest. The Raf– ERK system could equally well have been described as a PKC pathway had the series EGFR–Ras–Raf–ERK (as described in Chapter 12) not been discovered first. PKC is now better regarded as a booster
of the Ras–Raf–ERK pathway, but it could also have been the other way round, Ras being the booster of PKC–Raf–ERK.
581
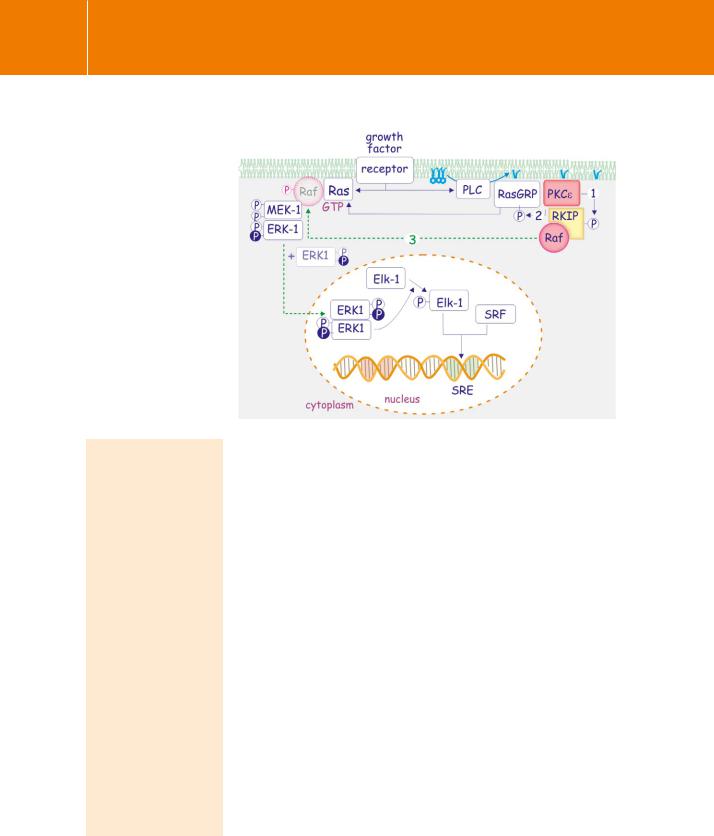
Signal Transduction
Fig 19.2 PKC causes activation of SRE via RKIP/RAF.
PKC and growth factors were initially thought to activate distinct signal transduction pathways resulting
in the activation of TRE and SRE respectively. However, PKC can activate SRE independently through phosphorylation of RKIP (1), an inhibitor of Raf. Phosphorylated RKIP loses its grip on Raf, which now associates with the Ras, MEK, ERK signalling cassette (3). Diacylglycerol also affects SRE through direct
activation of the Ras guanine exchange factor, RasGRP. This activation
is reinforced by PKCe-mediated phosphorylation (2), thus boosting the Ras-MEK-ERK pathway.
The S129 phosphorylation site on JNK. The S129 phosphorylation site is only slightly exposed
and would seem unable to interact directly with the upstream kinases MKK-4 or -7. Nor does
it appear to be able to affect the catalytic activity of JNK (according to the conformation observed in the non-phosphorylated JNK structure). However, binding of the scaffold protein JIP1 controls kinase activity and it is thus possible that pS129 changes this interaction and that the loss of
JIP1 binding allows for more efficient MKK4 or MKK7-mediated
phosphorylation of JNK34 (see page 463).
inhibitors showed that ERK and JNK are equal partners in maintaining cyclinD at an elevated level.30 However, the JNK pathway is not generally activated by mitogenic signalling, responding mainly to stress situations (see page 349).31 In some way, ERK provokes a strong JNK signal and this is where PKC comes into play.
Although the mitogenic pathway does not generally cause activation of c-Jun, it does induce its expression and stabilization. Enhancement of c-Jun is caused by phosphorylation and activation of CREB (see table 12.3, page 339). This involves the nuclear kinase MSK, a substrate of ERK1. Protection from the ubiquitylation/proteasome machinery is due to inactivation of GSK3 which otherwise phosphorylates c-Jun, making it a good substrate for the APC-ubiquitin ligase (see page 467). Despite low levels of activation,
increasing amounts of c-Jun lead to complex formation with ATF2 (forming an AP-1 complex) and together they bind the TRE-responsive element causing expression of RACK1. This binds to activated PKC and together they associate with JNK. The consequence is phosphorylation of JNK1. PKC , , and are likely candidates for this phosphorylation reaction, though other isoforms are not excluded. The phosphorylation enables subsequent phosphorylation and activation of JNK by the upstream kinases MKK4 or 7 to render JNK1 fully competent (Figure 19.3). Now things start to happen. The enhanced activity
of JNK1 together with increased expression of c-Jun leads to highly active c-Jun, which in turn boosts expression of yet more RACK1. A feed-forward amplification loop is established.
582
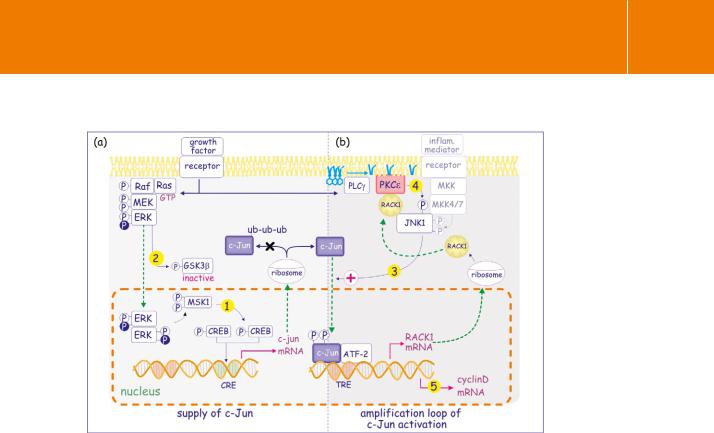
Protein Kinase C Revisited
Fig 19.3 PKC rewires the ERK pathway into an amplified JNK pathway.
(a) Growth factors activate the Ras–ERK pathway which leads to enhanced expression of c-Jun in two ways, through phosphorylation and activation of CREB (1) which enhances c-Jun transcription and through phosphorylation/inactivation of GSK3 (2). As a result, c-Jun is no longer phosphorylated and is protected against ubiquitylation.(b) Accumulation of c-Jun and residual activation levels of MKK4 or -7 lead to its
activation (3) and, after having joining up with ATF2, to increased expression of Rack1 mRNA. After translation, this attaches to PKC and together they associate with JNK1 leading to its phosphorylation (4). This renders the MKK-JNK1 signalling cassette much more efficient. An amplification loop results in an elevated c-Jun/ATF2- directed transcription of cyclinD (5) and shortens the G1 phase of the cell cycle.
Constant provision of c-Jun, due to a highly active Ras–ERK system, is a prerequisite for this pathway, as witnessed by its slow decline when cells are treated with inhibitors of the Ras–ERK pathway. Indeed, in melanoma cells this mode of amplification is only detectable when there are high levels of ERK activity. As a consequence, elevated levels of c-Jun lead to elevated
expression of cyclinD, again through a c-Jun/ATF2 complex which binds to its promoter. The c-Jun amplification loop explains, amongst many other things, how melanoma cells maintain such a high rate of proliferation. Not only this, elevated levels of RACK1 also protect against apoptosis32 and conversely, depletion of RACK1 sensitizes melanoma cells to UV-induced apoptosis and reduces their tumorigenicity. Alterations in RACK1 have been reported in different types of human tumours, with high levels of expression in non-small cell lung carcinoma and in colon carcinomas.33
583

Signal Transduction
Inflammatory mediators and cell transformation. We assume that both growth factor and inflammatory mediators are everpresent in the cellular environment. This is certainly the case for cancer cells. They often generate their own autocrine growth factors and these also elicit an inflammatory response with concomitant release of inflammatory mediators by surrounding cells and infiltrating blood-borne cells. This enhances their growth and dissemination because of the increased vascular permeability and general vascular remodelling around inflamed tissue.42
Over-expression of PKC isoforms and cellular transformation
Over-expression of PKC 1 in rat embryo (R6) fibroblasts induces the characteristic transformed cell phenotype (see page 306). The cells have a reduced requirement for growth factors and generate an autocrine mitogenic factor that possibly induces their own transformation.35 However, when injected into nude mice, these cells induce tumours with lower frequency and with a longer latent period than in cells transformed by the H-Ras oncogene. This should not be entirely unexpected, as phorbol esters act as tumour promoters or cocarcinogens, not carcinogens in their own right. Coexpression of H-Ras and PKC 1 certainly results in greatly enhanced transformation.36
In contrast, for R6 cells over-expressing PKC there is no tendency to transform.35 Instead, growth is retarded and the cells achieve lower saturation densities than normal. In glioma cells, PKC has a growth-promoting role, suppressing the expression of the cell cycle inhibitor p21WAF1/CIP1.37A summary of phenotypic changes caused by ectopic expression of members of the PKC family is presented in Table 19.3.
PKC has tumour suppressor qualities, actually causing v-Raf transformation of NIH-3T3 cells to revert.38 Likewise, PKC inhibits proliferation of vascular smooth muscle cells by preventing expression of cyclins D and E.39 It appears that, depending on the cells and on the circumstances, different isoforms of PKC, when over-expressed, can either induce or suppress the formation of the transformed cell phenotype.
Unfortunately, it has not been possible to draw any strong generalizations from these experiments. For sure, the tumour-promoting tendency of phorbol esters cannot be explained by a simple mechanism involving the activation of PKC. In addition, because prolonged treatment with phorbol esters has
the effect of down-regulating the expression of conventional and novel PKCs (a phenomenon often used to dissect their role in signal transduction), it
Table 19.3 Phenotypic changes caused by ectopic expression of different isoforms of PKC
PKC |
PKC |
PKC 1 |
PKC |
PKC |
PKC |
|
|
|
|
|
|
rat colonic |
glioma cells |
rat embryo |
vascular smooth |
rat fibroblasts and |
v-Raf |
epithelial cells |
|
fibroblasts (R6) |
muscle cells |
colonic epithelial |
transformed |
|
|
|
|
cells |
NIH-3T3 |
|
|
|
|
|
|
tumour |
tumour |
tumour |
tumour |
tumour |
tumour |
suppression |
promotion |
promotion |
suppression |
promotion |
suppression |
|
|
|
|
|
|
slower growth |
suppression |
growth in nude |
slower growth |
growth in nude |
reversion of |
low densities |
of p21WAF/CIP |
mice anchorage |
reduced |
mice anchorage |
transformed |
|
|
independence |
expression of |
independence |
phenotype |
|
|
synergy with |
cyclin D and E |
synergy with Ras |
|
|
|
Ha-Ras |
|
|
|
|
|
|
|
|
|
584

Protein Kinase C Revisited
is possible in some instances that the proliferative response may be due to absence of PKC rather than its activation. A case in point is PKC . This normally conveys an anti-proliferative and pro-apoptotic signal which is lost when its expression is down-regulated by prolonged treatment with PMA.40 Aberrant expression of PKC frequently occurs in cancers, with a general tendency for upregulation of PKC and down-regulation of PKC and PKC . In some cancers the correlation between expression of PKC isotypes and disease progression is striking, suggesting their potential uses as prognostic markers.41
Strong evidence against a crucial role of PKC in human tumorigenesis comes from the finding that few natural tumours have been found that express PKC mutations. There is just one lone example, a point mutation in PKC in a sub-population of highly invasive pituitary tumours and in thyroid follicular adenomas and carcinomas.43 This mutation alters the hinge region that links the regulatory and catalytic domains and, among other things, it causes a
selective loss of substrate recognition and an altered subcellular localization.44 How this explains aberrant growth regulation is not known.
Importantly, as already indicated, there are many other proteins that possess C1 domains, like those in the PKCs, that may also be regulated by phorbol esters. One of these, CalDAG-GEF (see page 234), is a guanine nucleotide exchange factor for Ras (and Rab), which promotes malignant transformation in fibroblasts when induced by phorbol ester.45
Regulation of cell polarity
Role of atypical PKC
The Par proteins: from Caenorhabditis elegans to mammals
The C. elegans par genes (par-1–6) were first identified in a genetic screen for maternal-effect mutations affecting the unequal partitioning of polar
granules to the posterior cell, during the asymmetric division of the single-cell embryo.46 Two of these genes, par-3 and par-6, encode PDZ domain proteins that colocalize at the anterior cortex by the end of prophase. At this point, Par-2, a Ring finger protein, and Par-1, an serine/threonine kinase, localize in a reciprocal manner to the posterior cortex (Figure 19.4). Par-4, another kinase, and Par-5, a 14-3-3 protein, are uniformly distributed throughout the cortex and
in the cytoplasm. The posterior Par-3–Par-6 complex is joined by the atypical PKC-3 and together, by guiding astral microtubules to the posterior site, they regulate the positioning of the mitotic spindle. A similar process occurs at the anterior site.47 The anterior and posterior are determined by the site of sperm entry. The microtubule organizing centre causes disruption of the actin–myosin network and Par-3, Par-6, and PKC-3 remain associated and are excluded from the entry point. This is the start of a series of events that enforces mutual exclusion of localization of the polarity complexes Par-3/6 and Par-1/2.48
585

Signal Transduction
Fig 19.4 Par in C. elegans.
(a) The sperm microtubule organizing centre arranges the disassembly of the actin cytoskeleton. This leads to a redistribution of Par proteins. Par-3 and Par-6 remain associated with the cytoskeleton and dominate the future anterior site. Par-1 and Par-2 remain ‘active’ at the future posterior site. Each Par complex has its specific kinase activity, Par-1 at the posterior and PKC-3 at the anterior site. This leads to phosphorylation of
other polarity-determining proteins and results in an asymmetric distribution of mRNA and proteins. (b) Then, following replication of DNA and during attachment of the chromosomes to the microtubule cytoskeleton, the Par complexes ensure the right orientation of the mitotic spindle by correctly positioning the astral microtubules.
Fig 19.5 Activation of a PKC-Par-6 by Cdc42.
(a) Activation of Cdc42, by a receptor binding an external polarity cue, leads to binding of Par-6/aPKC. This acts to relieve inhibition and allows phosphorylation and activation of the atypical PKC. The active complex now phosphorylates substrates that operate in the determination or maintenance of cell polarity. (b) Domain architecture of the Par-6 polarity complex. Domain interactions are indicated by red lines. (c) Structure of the domains involved in formation of the Par-6 polarity complex. The switch 1 and 2 regions of GTP-bound Cdc42 interact with the partial CRIB and with the PDZ domains of Par-6. Par-6 and PKC are linked through their PB1 domains (phox-bem domains, 1 and 2). Interacting surfaces are highlighted.
586