

Astrocyte Signaling in Neurological Disorders |
151 |
decarboxylase.43–45 In conditions such as ischemia, trauma, neurodegenerative disorders and hepatic encephalopathy, astrocytic glutamate uptake is impaired, potentially resulting in a disturbance of the glutamate-glutamine cycle, as well as an elevation in extracellular glutamate concentration that may lead to excitotoxic neuronal injury.46,47
9.1.2Responses of Astrocytes to Injury
There are three prototypical responses of astrocytes to CNS injury: reactive astrocytosis (astrogliosis), Alzheimer type II change and cell swelling. Reactive astrocytosis is the response of astrocytes to destructive injury, whereas the Alzheimer type II change is typically observed following metabolic/physiologic disorders. Cell swelling is an acute change that occurs in many forms of CNS insults and reflects a degenerative change resulting from impaired cell volume regulation.
9.1.2.1Reactive Astrocytosis
This response of astrocytes to destructive injury occurs robustly in acute CNS disorders such as stroke and trauma. Morphologically, reactive astrocytes display cytoplasmic enlargement, associated with the development of numerous thickened cytoplasmic processes48 (Figure 9.1). The nuclei are also enlarged, hyperchromatic and occasionally multi-nucleated. By electron microscopy the Golgi complexes are enlarged and increased numbers of mitochondria, ribosomes and glycogen granules are observed. Increased amounts of intermediate filaments (GFAP, vimentin and nestin) are a prominent feature of reactive astrocytes (Figure 9.2). All of these features are characteristic of metabolically activated cells. These changes are initially subtle at 3–4 days following injury and achieve their full expression at 2–3 weeks.48 A cluster of reactive astrocytes possessing numerous intertwining processes is often referred to as a ‘‘glial scar’’, although this scar bears no resemblance to the connective tissue scar seen in the rest of the body.
Reactive astrocytosis may also evolve in a more protracted fashion. This is the typical response in neurodegenerative disorders such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease and others. In contrast to the acute response, the reactive astrocytosis in these conditions tends to be less prominent.
a) Triggering Factors
While the precise factors responsible for the induction of reactive astrocytosis
are unclear, the release of proinflammatory cytokines such as IL-1, IL-2, IL-6, TNFa and IFN-g, likely from activated microglia, has been implicated.49–51
Elevated brain levels of these cytokines have been detected following CNS injuries,52 along with an over-expression of their receptors on astrocytes.53,54
Infusion of cytokines in brain has been shown to induce astrocytosis.49,50 Conversely, anti-inflammatory cytokines were shown to prevent astrocytosis.55 As noted above, astrocytes are well known to release many growth-promoting trophic factors, all of which have also been shown to stimulate astrocytosis,
152 |
Chapter 9 |
A B
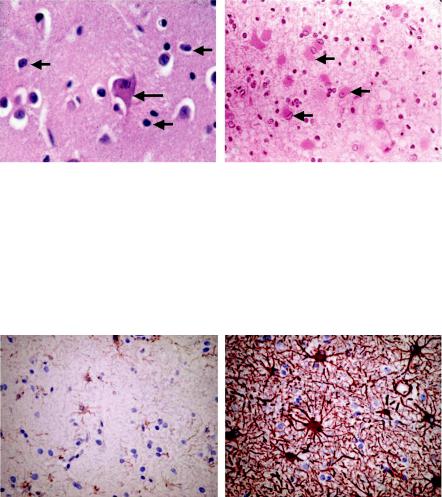
Figure 9.1 A Appearance of normal astrocytes (short arrows) from the striatum stained with hematoxylin and eosin (H&E). This figure discloses only the nuclei and little to no cytoplasm is evident. The large cell in the center of the field is a normal medium spiny neuron (long arrow). B Section of striatum from an individual who died with Huntington’s disease. Note the presence of numerous reactive astrocytes characterized by cells with abundant eosinophilic cytoplasm and nuclei that are larger and paler than normal (arrows).
A B
Figure 9.2 A GFAP-immunostained section of normal human cortex. Slight staining is observed in astrocytes. B GFAP-immunostained section of reactive astrocytes displays enhanced intensity of staining and additionally illustrates the increase in cytoplasmic volume and the extent and length of cytoplasmic processes. Both photographs were obtained from the same slide. The slide B image was just adjacent to a primary sarcoma of the brain, while the slide A image was approximately 1.5 cm away from the tumor where the tissue appeared normal.
possibly through autocrine e ect.56,57 Additionally, infusion of thrombin in brain or its addition to cultured astrocytes have also been shown to potently stimulate reactive astrocytosis.58,59
In addition to cytokines and thrombin, ATP also has been implicated as a stimulant for reactive astrocytosis. The P2Y receptor agonist ATP analog
Astrocyte Signaling in Neurological Disorders |
153 |
2-MeSATP has been shown to cause reactive astrocytosis.60–61 Microinfusion of 2-MeSATP in situ into the rat nucleus accumbens was shown to cause reactive astrocytosis, as demonstrated by upregulation of GFAP-immunoreactivity, cytoplasmic hypertrophy as well as an increase in the number of astrocytes.60 Reactive astrocytosis was also documented after injection of 2-MeSATP in the optic nerves of rats.61 However, as suggested by Franke et al. (1999),60 ATP may initially act on microglia resulting in their release of factors that ultimately cause reactive astrocytosis.
b) Factors Produced by Reactive Astrocytes
Reactive astrocytes are known to express and/or release macrophage antigen complex-1 (mac-1), growth factors (GDNF, BDNF, VEGF, NGF, CNF, bFGF, TGF-b) and antioxidant enzymes (superoxide dismutase, catalase and glutathione-s-transferase).48,62 Reactive astrocytes also increase the production of adhesion molecules, cytokines, chemokines and proteoglycans.63 Likewise, reac-
tive astrocytes have also been shown to over-express glutamate transporters GLAST64 and GLT1,65–67 the glucose transporter68,69 and gap junctional pro- teins.70–72 All of these factors provide the means for reducing oxidant injury,
promote repair and diminish the extent of excitotoxic neuronal/oligodendroglial injury.
Upregulation of ion channels such as the inward rectifier K1 channel Kir2.3 and
L-type Ca21 channels were also observed in reactive astrocytes in di erent neurological conditions.73–75 While the significance of the upregulation of these ion
channels is unclear, it has been proposed that they may contribute to the maintenance of ion homeostasis and to enhance the release of neurotrophic factors.74,76
c) Functions of Reactive Astrocytes
The functional significance of reactive astrocytosis remains unclear. As noted above, it has been proposed that reactive astrocytes are involved in restoring the integrity of the blood-brain barrier, maintaining appropriate extracellular glu-
tamate levels, supporting neurite outgrowth and providing neurotropic factors (NGF, CNTF, bFGF).48,77
While beneficial functions have been proposed for reactive astrocytes, these cells have also been implicated in detrimental outcomes. Thus, glial scar formation may be an impediment to axonal regeneration.78 This aspect will be elaborated upon below.
It should be emphasized that reactive astrocytes may be heterogeneous depending on the age of the disease process (acute or chronic), location of the astrocytes relative to the core of the lesion and the nature of the disease process the astrocyte is responding to. This heterogeneity needs to be considered when interpreting conflicting views on the significance of the reactive astrocytes following CNS injury. Additionally, many conclusions regarding their significance have been reached based on cell culture models, whose fidelity to the in vivo condition is frequently uncertain.
154 |
Chapter 9 |
9.1.2.2Cell Swelling
Astrocyte swelling is a common response to CNS injury. It is often observed after various intoxications (e.g. dinitrophenol, triethyltin, hexachlorophene, iso-
niazid),79 Reye’s syndrome,80–82 acute hepatic encephalopathy (acute HE),83–84 as well as in the early phase of ischemic stroke85–87 and traumatic brain injury (TBI).88–89 A major consequence of such cell swelling is the development of
cytotoxic edema, a life-threatening process that may lead to increased intracranial pressure and brain herniation. Swollen astrocytes often display morphologic abnormalities (Figure 9.3), suggesting that such swelling may also lead to astrocyte dysfunction and an inability of these cells to carry out their vital functions.
Two forms of brain edema are recognized: cytotoxic and vasogenic.90 Cytotoxic edema is largely due to astrocyte swelling, and the signaling systems involved in such swelling will be discussed below. Vasogenic edema is due to an extracellular fluid accumulation due to a breakdown of the blood-brain barrier. A discussion of signaling systems involved in vasogenic edema is beyond the scope of this review.
9.1.2.3Alzheimer Type II Astrocytosis
In chronic metabolic disorders such as hepatic encephalopathy, hyperammonemia, Wilson’s disease, renal failure, endocrine disorders and other conditions, astrocyte nuclei become enlarged and pale and no cytoplasmic abnormalities are observed by light microscopy, except for the presence of lipofuscin pigment (often a marker of oxidative stress) (Figure 9.4). Such cells
A B
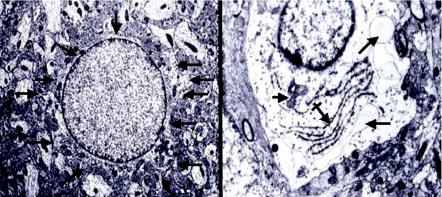
Figure 9.3 A Electron micrograph of a cortical astrocyte from a normal rat. The cytoplasm is barely discernable in this figure (the margins of the cytoplasm are outlined by arrows). B Astrocyte from a rat with acute hepatic encephalopathy showing a marked expansion of the cytoplasm which is clear (‘‘watery’’) and displays several vacuoles (long arrows), slightly disrupted endoplasmic reticulum (crossed arrow) and an irregular-appearing mitochondrion (short arrow).