

Regulation of Muscle Proteostasis via Extramuscular Signals |
79 |
protein structures that make up the contractile apparatus prior to tra cking of the disassembled proteins to the proteasome or lysosome for final destruction.4
5.2The E ects of Extramuscular Signals on Global Proteostasis in Muscle
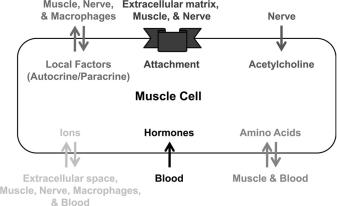
A host of extramuscular signals that regulate proteostasis have been identified both in man and in biomedical models such as cultured muscles, rodents and invertebrates (Figure 5.1). The next sections will describe the role of some of these signals in the regulation of muscle proteostasis.5,6 However, it is important to state that classifying extracellular signals as simply either anabolic or catabolic per se is an oversimplification since signals may be anabolic under some conditions and catabolic in others, probably as a result of crosstalk with other signals. Furthermore, detailed mechanisms by which many extracellular signals regulate muscle proteostasis are as yet unavailable, which highlights the paucity of research into this area. Nevertheless these signals still warrant highlighting as they are likely to emerge as important in the regulation of muscle proteostasis.
5.2.1Hormones
One class of signals is hormones, which are produced by endocrine organs at a distal site from muscle. These are thought to generally promote catabolism (e.g. glucocorticoids, thyroid hormone (TH)) or anabolism (e.g. insulin, testosterone) and in many cases are directly responsible for muscle wasting in patients su ering from endocrine disorders.
Insulin has been suggested to control 50% of muscle size during development of rodents and therefore would be expected to play a major role in human beings. Indeed insulin increases muscle protein synthesis and suppresses degradation (see Section 5.2.2.1 for detailed discussion), demonstrating its importance in
Figure 5.1 Extramuscular signals that muscle integrates to regulate proteostasis.
80 |
Chapter 5 |
muscle proteostasis. In contrast, glucocorticoids (i.e. cortisol) decrease both protein synthesis and degradation, as does epinephrine, while TH appears to promote both processes. Presumably when both processes are inhibited or promoted the net e ect will depend upon the quantitative extent of inhibition or activation. For example, TH promotes degradation to a larger extent than synthesis, which results in a net shift toward catabolism of muscle protein. Finally, despite its potent e ects in developing/deficient states, growth hormone (GH) in healthy adults has no anabolic e ects on skeletal muscle (despite increases in insulin-like growth factor (IGF-1) expression), instead increasing tendon collagen synthesis.
5.2.1.1Mechanisms of Action: Glucocorticoids
Muscle wasting is observed in patients with excessive levels of glucocorticoids (i.e. Cushing’s syndrome).7 In addition, glucocorticoid concentrations are increased in cases of sepsis and burns when muscle is also undergoing atrophy. However, little is known of precisely how glucocorticoids, for example endogenous cortisol and clinically used dexamethasone, cause changes in proteostasis in humans. Glucocorticoids are traditionally thought of as binding to intracellular hormone receptors, which in turn bind to glucocorticoid response elements in DNA, thereby causing altered expression of mRNA. Indeed, current evidence suggests that glucocorticoids induce expression of a ubiquitin E3 ligase and proteasome activator subunit, which may contribute to increased degradation that is observed. On the other hand, glucocorticoid treatment also represses the expression of the myogenic transcription factors, myoD and myogenin, which presumably results in decreased expression of muscle-specific genes, including myosin. Additionally, glucocorticoids increase the expression of 4E binding protein 1 (4E-BP1), which is a translational repressor, thus reducing translational e ciency. Glucocorticoids also reduce the transcription of insulin receptor substrate (IRS), phosphoinositide 3 kinase (PI3K) and protein kinase B (Akt) (see Section 5.2.2.1 for more on these signals). Thus, glucocorticoids are capable of changing the responsiveness of muscle to other extramuscular signals known to regulate proteostasis. These observations demonstrate negative regulation over multiple levels of protein synthesis.
5.2.1.2Mechanisms of Action: TH (T3)
Muscle wasting is observed in patients with excessive levels of TH (i.e. thyroid toxicosis).8,9 Indeed, TH has a well-described catabolic e ect on adult skeletal muscle and, as mentioned above, sustained elevations in blood thyroid hormone levels result in muscle weakness and wasting. Like glucocorticoids, TH is thought to influence muscle proteostasis via binding to intracellular hormone receptors (TH receptors), which in turn bind to TH response elements in DNA and thus cause changes in mRNA expression. TH exposure induces expression of the lysosomal enzyme cathepsin C and multiple subunits of the proteasome, which fits with its purported role in protein degradation. Similarly,
Regulation of Muscle Proteostasis via Extramuscular Signals |
81 |
TH also increases the expression of the synthetic apparatus, notably increasing the expression of several transcription factors including myogenic factor 6 (Herculin), several ribosomal proteins and translation initiation factors. Notably, TH also increases the transcription of cellular receptors for muscle growth factors and also the transcription of intramuscular signaling proteins, thus TH is capable of changing the responsiveness of muscle to other extramuscular signals known to regulate proteostasis. Nevertheless, TH-induced catabolism must prevail over anabolism for muscle atrophy to occur (at least independent of other signal interactions).
5.2.1.3Mechanisms of Action: Testosterone
Testosterone is a well-known anabolic hormone, the administration of which increases the basal rate of muscle synthesis in both healthy and hypogonadal young men.10,11 Conversely, testosterone concentrations decline with aging, perhaps consonant with a role in sarcopenia (at least in men). Like glucocorticoids and TH, testosterone is thought to influence muscle proteostasis via binding to intracellular hormone receptors (androgen receptors), which in turn bind to androgen response elements in DNA and thus cause changes in mRNA expression. Notably IGF-1 expression increases in response to testosterone treatment, thus suggesting that the reason for similar e ects of testosterone and IGF-1 on muscle proteostasis is that IGF-1 acts downstream of testosterone. In support of this, testosterone increases the phosphorylation of mTOR and its downstream targets 40S ribosomal protein S6 kinase 1 (p70S6K1) and 4E-BP1, which are parts of the IGF pathway, thereby increasing translational e ciency.
Additionally, testosterone has been show to bind to receptors at the muscle plasma membrane. These receptors are G-protein coupled receptors. Unlike insulin receptors, G-protein coupled receptors are not themselves kinases. However, like insulin receptors, G-protein coupled receptors work by recruiting other proteins, in this case G-proteins, to the receptor when bound to ligand. Upon activation of the receptor a G-protein in turn activates phospholipase C, which produces inositol trisphosphate (IP3), which then acts upon IP3 receptors located at the sarcoplasmic reticulum to stimulate release of intramuscular Ca21 with an oscillatory pattern. The intramuscular Ca21 can then, perhaps, act like Ca21 release in response to contraction and also activate the Ras regulated protein kinase cascade, which can upregulate initiation/ elongation factor expression and as such the capacity for protein synthesis. Alternatively or in addition, Ca21 could activate the calmodulin-activated phosphatase calcineurin, which mediates hypertrophic response through regulation of downstream nuclear factor of activated T cells (NFAT) transcription factor and thus initiates a hypertrophic gene program.
5.2.1.4Mechanisms of Action: Epinephrine
Adrenal-derived epinephrine is found elevated in a rare endocrine disorder associated with muscle wasting: pheochromocytoma.12 More commonly,