

Posob_2012_Ok
.pdfµi′′< µi′, то dni′′ > 0, т.е. компонент переходит из второй фазы в первую, если его химический потенциал в этой фазе больше, чем в первой; если же химический потенциал компонента во второй фазе меньше, чем в первой, компонент будет переходить из первой фазы во вторую. Таким образом, компонент самопроизвольно переходит из фазы, в которой его химический потенциал больше, в фазу, в которой химический потенциал меньше. Этот переход продолжается до выравнивания химических потенциалов компонента в обеих фазах.
2.4. Правило фаз, его вывод и применение к классификации систем
Правило фаз оперирует с основными понятиями о компоненте, фазе, числе степеней свободы (вариантности системы). Понятия фазы и компонента определены выше; остается определить понятие о числе степеней свободы системы.
Состояние системы характеризуется некоторыми величинами - параметрами (давление, удельный объем, температура, концентрация).
Если дана конкретная система, то не все эти параметры можно выбрать произвольно. Например, если вода находится в равновесии со своим паром, то, выбрав произвольную температуру, мы лишаемся возможности произвольного выбора давления и наоборот.
Под термодинамическими степенями свободы, или просто степенями свободы, подразумеваются независимые параметры системы, находящейся в термодинамическом равновесии, которые могут принимать произвольные значения в определенном интервале, причем число фаз не изменяется. Другими словами, степенями свободы называются те параметры системы, которые играют роль независимых переменных. Все остальные параметры будут их функциями. Количество степеней свободы называется также вариантностью системы (В).
Пусть в некоторой системе К - число компонентов, Ф - число фаз. Номер компонента будем ставить индексом внизу, номер фазы индексом вверху. В качестве независимых переменных принимаем Р, Т и К-1 концентрацию. Поскольку система находится в равновесии, температура, давление и химические потенциалы компонентов во всех фазах равны между собой.
Так как количество независимых концентраций для каждой системы равно К-1, а количество фаз равно Ф, то общее количество независимых концентраций в системе равно (К-1)Ф, а общее количество независимых переменных в системе на 2 больше (давление и температура) и равно (К- 1)Ф +2.
Выясним теперь общее количество уравнений связывающих эти независимые переменные. Учитывая равенство химических потенциалов каждого компонента во всех фазах, можно записать: для первого компонента:
31
µ11 = µ12 = µ13 = ... = µ1ф. Для второго компонента: µ21 = µ22 = µ23 = ... µ2ф . . .
. µK1 = µK2 = µK3 = ... µKф. Количество уравнений в строке равно Ф-1, количество строк равно К, тогда 2 + Ф(К - 1) независимых переменных связывает (Ф-1)К уравнений. Отсюда следует, что не все 2 + Ф(К -1) независимых переменных являются таковыми на самом деле. Из математики известно, что если количество переменных в системе больше числа уравнений, то количество независимых переменных равно количеству всех переменных за вычетом количества уравнений, их связывающих: В = 2 + Ф(К-
-1) -К(Ф -1) = 2 + ФК - Ф - КФ +К = К - Ф + 2, |
|
В + Ф = К + 2. |
(2.26) |
Это и есть правило фаз: в изолированной системе число фаз + число степеней свободы равно числу компонентов + 2.
Если какой-нибудь параметр, характеризующий систему, принима-
ется постоянным ( Т = const или Р = const), то |
|
В + Ф = К + 1. |
(2.27) |
А если постоянными поддерживаются и давление, и температура - |
|
правило фаз принимает вид: |
|
В + Ф = К. |
(2.28) |
3.Однокомпонентные системы
3.1.Диаграмма состояния однокомпонентной системы
Применим правило фаз (В + Ф = К + 2) к однокомпонентным системам для случая, когда давление и температура могут изменяться:
В = 1 +2 - Ф, если Ф = 1, то В = 3 - 1 = 2 - дивариантное равновесие (Ф может быть жидкой (L), твердой (S) или газообразной (V) фазой);
Ф= 2 В = 3 - 2 = 1 - моновариантное равновесие (соответствующи-
ми фазовыми равновесиями могут быть: L1 + L2 (равновесие двух несмешивающихся жидких фаз); L + V; S1 + S2 (равновесие двух аллотропных твердых фаз); S + L; S+V. Равновесие L1 + L2 в однокомпонентных системах до настоящего времени экспериментально не наблюдалось);
Ф= 3 В = 3 - 3 = 0 - нонвариантное равновесие (соответствующими фазовыми равновесиями могут быть: S1 + S2 + S3 (равновесие трех алло-
тропных твердых фаз); S1 + S2 + L; S1 + S2 + V; S + L1 + L2; S + V + L). Рав-
новесие S + L1 + L2 в однокомпонентных системах экспериментально не обнаружено.
Рассмотрим теперь диаграммы состояния однокомпонентных сис-
тем.
Если отложить по оси абсцисс температуру, а по оси ординат - давление, то получится диаграмма состояния, дающая графическое изображение состояния однокомпонентной системы.
Состояние такой системы определяется точкой, отвечающей опре-
32
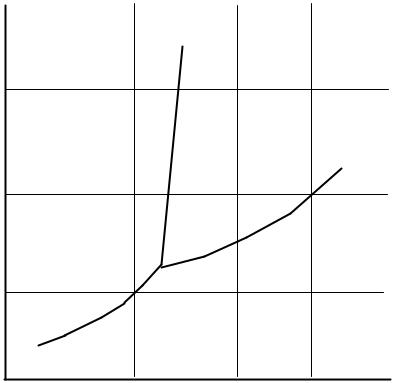
деленным температуре и давлению. Такая точка называется фигуративной точкой этой системы. Диаграмма состояния приведена на рис. 3.1.
На рис. 3.1 нонвариантному равновесию (определенные давление и температура) отвечает точка А - тройная точка. Моновариантные равновесия - непрерывные линии, исходящие из точки А: АВ; АС; АD. Дивариантным равновесиям отвечают участки площади - поля диаграммы: ВАD - твердое; DАС - жидкое; ВАС - парообразное. Каждой фазе отвечает определенный геометрический образ: поле, линия, точка. Изобарические процессы изображаются на диаграмме состояния горизонтальными линиями, параллельными оси температур (Р1; Р2; Р3). Изотермические процессы изображаются вертикальными линиями (Т1; Т2; Т3).
P |
|
|
|
|
D |
|
|
P3 |
|
|
|
P2 |
|
|
C |
|
|
|
|
P1 |
A |
|
|
B |
|
|
|
T1 |
T2 |
T3 |
T |
Рис. 3.1. Диаграмма состояния однокомпонентной системы |
|||
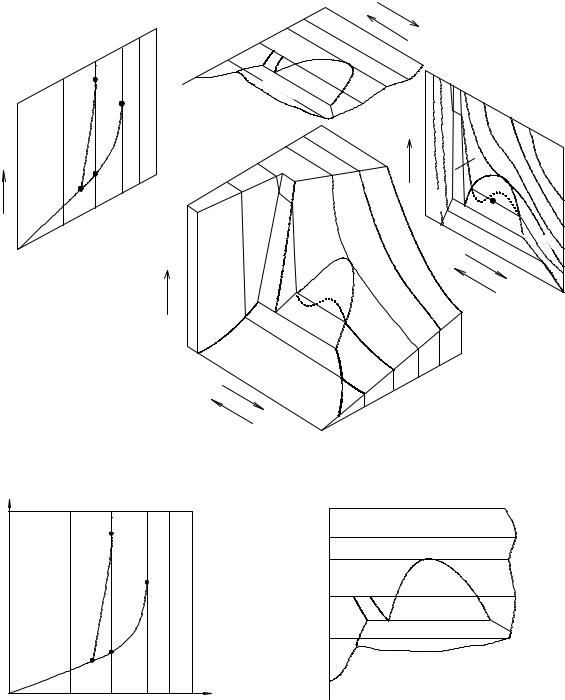
Чтобы изобразить графически соотношения между Т, Р, V, необходимо использовать трехмерную систему координат. Нанося экспериментальные данные о нонвариантных и моновариантных равновесиях ( значения Т, Р, V), получим полную диаграмму состояния.
В качестве примера на рис. 3. 2 приведем полную диаграмму состояния СО2.
33
|
|
|
|
|
|
|
|
|
|
|
|
4 |
|
|
5 T |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2 |
|
3 |
|
|
|
C |
V |
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
1 |
|
|
|
|
|
|
|
|
|
|
0 1 |
|
|
|
|
|
|
|
a |
|
|
|
|
0 |
|
|
|
L+ |
|
|
|
|
2 3 |
|
|
|
|
|||
|
|
|
|
|
|
S |
+ |
V |
|
|
|
|
4 |
|
|
||||||||
|
|
L |
K |
|
|
|
|
|
V |
|
|
|
|
|
|
|
|
5 |
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
4 5 |
|
|
|
|
|
+ L |
|
|
|||||
|
|
|
|
|
|
|
|
|
|
3 |
|
|
|
P |
|
|
|
|
|||||
P |
S |
|
|
|
|
T |
2 |
|
|
|
|
|
|
|
|
|
|
|
S |
|
K |
||
|
V |
|
|
1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
4 |
5 |
|
|
a' |
|
|
|
|
|
|
|
|
|
|
|
|
|
• |
|||
|
|
c O |
3 |
0 |
|
|
|
a'' |
|
|
|
|
|
S |
|
|
L |
+ |
|
||||
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
V |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
S |
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
+V |
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
K V |
|
|
|
|||||
|
0 |
|
|
|
P |
|
|
|
|
|
|
|
|
|
|
C |
|
||||||
|
|
|
|
S |
L |
|
|
L |
|
|
• |
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
V |
|||||||
|
|
|
|
|
|
|
O' |
+ |
|
b'• |
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
c' • • |
|
• |
|
|
L |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
S |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O'' |
|
|
+ |
V |
•b'' |
V |
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
• O''' |
|
|
|
T |
|
|
|
|
|
|
|
|
|
|
S |
+V |
|
|
|
|
|
|
5 |
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
•c'' |
|
4 |
|
|
|
|
||||||
|
|
|
|
|
|
|
C |
|
|
|
|
|
3 |
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
V |
|
|
|
|
|
|
1 |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
0
Рис. 3.2. Полная диаграмма состояния СО2
P |
|
|
|
|
|
|
|
|
|
|
a |
|
|
|
|
|
|
|
|
|
K |
|
|
|
S |
|
L |
|
|
|
|
|
|
|
|
|
|
|
|
|
c• |
O |
b V |
|
|
|
|
0 |
1 |
|
2 |
3 |
4 |
5 |
T |
|
|
||||||
|
Рис. 3.3. РТдиаграмма |
|
|||||
T
5 |
|
|
|
|
V |
|
|
|
|
|
|
||
4 |
|
|
|
|
• K |
|
3 |
|
|
|
|
||
|
a' |
a" |
L |
|
L + V |
|
2 |
• |
• |
||||
• |
• |
|
||||
|
O' • |
• |
b' |
b'' |
||
|
|
•O''' |
||||
1 |
•c' |
O'' |
S + V |
|||
0 |
|
|
|
|
|
|
Рис. 3.4. TVдиаграмма
V
Проецирование полной диаграммы состояния на координатные плоскости (РV, VТ, ТР) позволяет получить различные проекции - так называемые РТ-диаграммы, VТ-диаграммы (см. рис. 3. 4) и РV-диаграммы
(см. рис. 3. 5).
Наиболее удобны и широко применимы РТ-диаграммы.
34

P |
0 |
1 |
2 |
3 |
4 |
5 |
6 |
a' a"
L K
b' |
L + V |
b'' V |
|
|
|
O' O'' |
|
O''' |
c' |
S + V |
c'' |
|
V
Рис. 3.5. PVдиаграмма
3.2.Уравнение Клапейрона-Клаузиуса
Всистеме, состоящей из нескольких фаз чистого вещества, находящихся в равновесии, возможны переходы вещества из одной фазы в другую. Такие переходы называются фазовыми переходами или превращениями агрегатных состояний.
Рассмотрим равновесный переход одного моля вещества из одной фазы (1) в другую (2), совершающийся при постоянных давлении и температуре. Изменение внутренней энергии системы равно (производится лишь
работа расширения): U2 - U1 = T(S2 - S1) - P(V2 - V1), откуда U2 - ТS2 + PV2 = U1 - TS1 + PV1, или G2 = G1, т.е. изобарные потенциалы единицы массы чистого вещества в двух фазах, находящихся в равновесии, равны между собой. Изохорные потенциалы двух равновесных фаз не равны между собой, причем их разность равна максимальной работе процесса перехода: F2
-F1 = -Аmax = -P(V2 - V1).
Напишем уравнения полных дифференциалов для изобарных потенциалов одного моля чистого вещества в двух равновесных фазах 1 и 2: dG2 = V2dP - S2dТ; dG1 = V1dp - S1dТ. Вычитая второе уравнение из перво-
го, получим: dG2 -dG1 = (V2- - V1)dP - (S2 - S1)dT Изменения p и T здесь бы-
ли не независимыми, а такими, при которых сохранялось равновесие между фазами 1 и 2. Таким образом, между P и Т сохранялась функциональная связь, соответствующая фазовому равновесию. Если G1 = G2 (равновесие при давлении p и температуре Т), то G1 +dG1 = G2 + dG2 (равновесие при
35
давлении P + dP и температуре Т + dТ), т.е. dG1 = dG2, или dG2 - - dG1 = 0.
Итак, (V2 - V1)dP - (S2 - S1)dТ = 0, или |
|
dP/dТ = (S2 - S1) / (V2 - V1). |
(3.1) |
Взаимное превращение фаз рассматривалось здесь как равновесное |
|
и изотермическое, поэтому: |
|
S2 - S1 = Q/Т = ∆HФ. П./ТФ. П. |
(3.2) |
Здесь ∆HФ. П. - теплота фазового превращения, поглощаемая при переходе моля вещества из фазы 1 в фазу 2; V2 - V1 - разность мольных объемов двух фаз. Из уравнений (3.1) и (3.2) получим: dP/dТ = ∆H/(Т(V2-V1)),
или |
|
∆H = Т(V2-V1)dP/dТ. |
(3.3) |
Уравнение (3.3) называется уравнением Клапейрона-Клаузиуса и является общим термодинамическим уравнением, приложимым ко всем фазовым переходам чистых веществ, т.е. к превращениям агрегатных состояний.
3.3. Фазовые переходы 1-го рода
Фазовые переходы, характеризующиеся равенством изобарных потенциалов двух сосуществующих в равновесии фаз и скачкообразным изменением энтропии и объема при переходе вещества из одной фазы в другую, называются фазовыми переходами первого рода. К ним относятся агрегатные превращения - плавление, испарение, возгонка и др.
Рассмотрим подробнее процессы плавления, испарения и возгонки, представляющие наиболее общий интерес.
3.3.1. Плавление
Теплота плавления всегда положительна. Мольный объем (а также и удельный) жидкой фазы (V2) в общем случае может быть больше или меньше объема твердой фазы (V1). Отсюда в соответствии с (3.3) вытекает, что величина dP/dТ или обратная ей величина dТ/dP, характеризующая изменение температуры с увеличением давления, может быть положительной или отрицательной:
dP/dТ = ∆H/Т(V2-V1); dТ/dP = Т(V2-V1)/∆H.
Это значит, что температура плавления может повышаться или понижаться с увеличением давления. Если ∆V < 0, то при увеличении давления в системе температура плавления вещества уменьшается. Если же ∆V > 0, то при увеличении давления в системе температура плавления вещества увеличивается.
Эти выводы вытекают из принципа Ле-Шателье. Увеличение Р ведет к процессу с уменьшением объема или увеличением плотности. Уменьшение Р ведет к процессу с увеличением объема или уменьшением плотности. Увеличение Т ведет к процессу с поглощением тепла. Уменьшение Т ведет к процессу с выделением тепла.
36
Величина dТ/dP положительна для огромного большинства веществ. Она имеет отрицательное значение лишь для воды, висмута и немногих других веществ, для которых плотность жидкости при температуре плавления больше плотности твердой фазы и, следовательно, VЖ - VТ < 0.
На диаграммах состояния однокомпонентных систем кривые плавления проходят наиболее круто, отклоняясь от вертикали вправо или влево в зависимости от изменения мольного или удельного объемов равновесных твердой и жидкой фаз.
3.3.2. Испарение и возгонка
Теплоты испарения и возгонки - перехода жидкой или твердой фазы в газообразную - положительны. В этих случаях всегда мольные (или удельные) объемы газообразных фаз значительно больше соответствующих объемов жидкой или твердой фаз, т.е. dP/dТ, а значит и dТ/dP всегда положительны. Следовательно, температуры испарения и возгонки всегда повышаются с ростом давления.
При температурах, далеких от критических, V2>>V1, поэтому
∆H = Т(dP/dТ)VГ . |
(3.4) |
Кривая испарения на диаграммах |
состояния однокомпонентных |
систем ограничена сверху критической точкой, в которой исчезает различие между жидкостью и паром. Жидкость может быть охлаждена ниже температуры плавления, кривая пара в этом случае является частью общей кривой.
Кривая возгонки (сублимации), или кривая давления пара твердого вещества, изображающая графически моновариантное равновесие твердого тела с его паром, имеет вид, сходный с кривой испарения. Снизу ограничена абсциссой 0 К, сверху - тройной точкой.
Кривая возгонки имеет больший угловой коэффициент наклона, чем кривая испарения, так как величина dР/dТ из уравнения (3.4) будет определяться величиной теплового эффекта фазового перехода, которая у процесса возгонки всегда больше, поскольку равняется сумме теплот плавления и испарения.
Если к тому же вдали от критической температуры насыщенный
пар можно считать подчиняющимся законам идеальных газов, тогда |
|
VГ = RT/P, ∆H = RТ2 dlnP/dТ, или |
(3.5) |
dlnP = ∆HdТ/(RТ2). |
(3.6) |
Теплота испарения жидкостей изменяется с температурой, не сильно убывая при средних температурах и очень сильно - вблизи критической температуры, при которой ∆H = 0.
Зависимость давления насыщенного пара от температуры дает интегрирование уравнения Клапейрона-Клаузиуса.
Давление насыщенного пара жидкости резко возрастает с повыше-
37
нием температуры, о чем свидетельствует опыт. Функциональная зависимость давления насыщенного пара жидкости (и твердых веществ) от температуры может быть выражена уравнением (3.3), а вдали от критической температуры - уравнением (3.6).
Считая теплоту испарения (возгонки) постоянной в небольшом интервале температур, можно легко проинтегрировать последнее уравнение:
P2 |
∆H T 2 |
dT |
; |
ln |
P |
= |
∆H(T −T ) |
. |
(3.7) |
|
∫d ln P = |
R ∫ |
T2 |
P |
RT T |
1 |
|||||
|
|
|
|
|
2 |
|
2 |
|
|
|
P |
T1 |
|
|
|
1 |
|
1 2 |
|
|
|
1 |
|
|
|
|
|
|
|
|
|
|
Формула (3.7) |
позволяет |
вычислять скрытую теплоту испарения |
||||||||
(сублимации), если известны давления пара при двух разных температурах, или же вычислять давления паров, если известна скрытая теплота и давление пара при одной какой-либо температуре. Дифференциальное уравнение dln p = ∆HdT/(RТ2) можно интегрировать и с помощью неопределенных интегралов:
lnP = - ∆H/RT + const. |
(3.8) |
В соответствии с этим зависимость давления насыщенного пара жидкости (или кристаллического вещества) от температуры может быть выражена прямой линией в координатах lnP - 1/Т (в этом случае тангенс угла наклона прямой равен (-∆H/R), откуда легко находится ∆H. Эта зависимость имеет место лишь в некотором интервале температур, далеких от критической.
Однако уравнение (3.8) не охватывает зависимости давления насыщенного пара от температуры во всем интервале температур - от температуры плавления до критической. С одной стороны, теплота испарения зависит от температуры и интегрирование должно производиться с учетом этого. С другой стороны, насыщенный пар при высоких температурах нельзя считать идеальным газом. Поэтому уравнение, охватывающее зависимость p = f(Т) в широком интервале температур, неизбежно становится эмпирическим.
Такое уравнение можно получить путем применения уравнения
Кирхгофа к процессу парообразования. По Кирхгофу: |
|
d∆H / dТ = СНАС. ПАР - СЖ ∆СP |
(3.9) |
(знак ставится потому, что при высоких давлениях замена СНАС. ПАР на СP является грубым допущением). Интегрируя выражение (3.9), получим:
∆H |
T |
T |
|
∫d∆H = ∫∆CpdT ; |
∆H = ∆H0 + ∫∆CpdT . |
(3.10) |
|
∆H0 |
0 |
0 |
|
Здесь |
∆H0 - постоянная величина, получаемая экстраполяцией кри- |
||
вой ∆H= f(Т) до 0 К.
Подставим это соотношение (3.10) в уравнение dlnP = (∆H/RТ2)dТ:
38
dlnP = ∆H0dТ/(RT2) + (0∫T ∆CPdТ)dТ/(RT2), и ∫ dlnP = (∆H0 ∫dТ/Т2)/R + (1/R)∫ (0∫T ∆CPdТ/Т2)dТ, откуда:
ln p = (-∆H0/R)(1/Т) + (1/R)∫(0∫T ∆CPdТ/Т2)dТ + j, (3.11)
где j - константа интегрирования.
Константы ∆H0 и j могут принимать различные значения в зависимости от точности используемых данных по СP. Если же СP для пара и жидкости известны точно и в широком интервале температур, то j определяется однозначно и называется истинной химической постоянной.
Можно несколько иначе подойти к решению вопроса. Учитывая, что теплоемкости являются функцией от температуры и эта функциональная зависимость представляется степенными рядами, после решения уравнения Кирхгофа получим следующее соотношение: ∆H = ∆H0 + aТ + bТ2 + сТ3 +..., тогда
dlnP=∆H0dТ/(RT2) + (а/R)(dТ/Т) + (b/R)dТ + (с/R)ТdТ +... .
Интегрируем: ln p = - ∆H0/(RТ) + (а/R)(lnТ) + (b/R)(Т) + .... +const,
или
lg p = -∆H0 /(4,575Т) + а/1,987(lgТ)+ bТ/4.575 + ... + const'.
Нернст упрощает это уравнение тем, что принимает а/1,987 = =7/(2·2) =1,75; он ограничивается только одним членом с Т после логарифмического члена и вводит обозначение - ε вместо b, тогда
lg p = -∆H0/(4.575Т) + 1.75lgТ - εТ/4.575 + i, (3.12)
где величины ∆H0, ε и i подбираются эмпирически. Величина i называется условной химической постоянной. Эта величина для многих веществ с двух- и многоатомными молекулами близка к трем, если давление выражено в атмосферах.
Уравнение Нернста (3.12) является менее точным уравнением, нежели уравнение (3.11).
3.3.3. Тройная точка
Тройная точка - графическое изображение нонвариантного процесса (равновесие трех фаз в однокомпонентной системе). Р и Т имеют для каждого вещества строго определенные значения.
Н2О : Р = 4,6 мм рт ст; Т = +0,098 0С, СО2 : Р = 5,1 мм рт ст; Т = -56,6 0С.
3.4. Полиморфизм, энантиотропия, монотропия
Полиморфизм - возможность существования вещества в различных кристаллических модификациях.
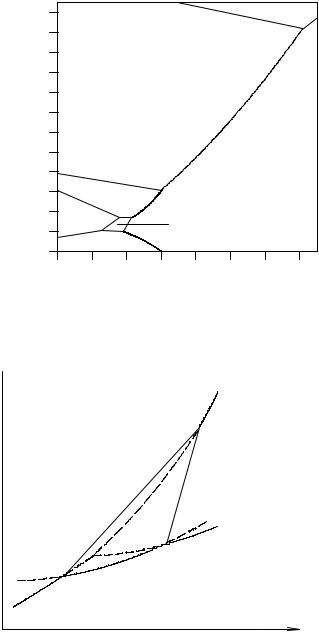
Примером диаграммы состояния с большим количеством полиморфных превращений является диаграмма состояния воды (см. рис. 3. 6).
39
P,атм
24000 |
|
|
|
|
|
|
|
VII |
O: S(I)+L+V, +0.008°C, 4.6 |
22000 |
|
|
|
|
|
|
|
G |
мм. рт. ст |
20000 |
|
|
|
|
|
|
|
|
C: S(I)+L+S(III), -22°C, 2115 |
18000 |
|
|
VI |
|
|
|
|
ат. |
|
16000 |
|
|
|
|
|
|
D: S(III)+L+S(V), -17°C, 3530 |
||
|
|
|
|
|
|
|
|
||
14000 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ат. |
|
12000 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
E: S(V)+S(VI)+L, +0.16°C, |
|
10000 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
L |
|
6380 ат. |
|
8000 |
|
|
|
E |
|
|
|
||
6000 |
|
V |
|
|
|
|
|
G:S(VII)+L+S(VI), +81.6°C, |
|
|
S D |
|
|
|
|
|
|||
4000 |
II |
F |
III |
|
|
|
|
22400 ат. |
|
2000 |
I C |
|
|
|
|
F: S(I)+S(II)+S(III), -34.7°C, |
|||
|
|
O |
|
|
|
|
|||
0 |
|
|
|
|
|
|
2170 ат. |
||
|
|
|
|
|
|
|
|
||
-60 |
-40 |
-20 |
0 |
20 |
40 |
60 |
80 |
S: S(II)+S(III)+S(V), -24.3°C, |
|
|
|
|
|
|
t,°C |
|
|
|
3510 ат. |
|
|
|
|
Рис. 3. 6. Диаграмма состояния воды |
|||||
P 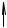
|
|
|
•B(151°C,131МПа) |
|
|
Sр |
Sм |
L |
|
|
|
|
||
D |
O |
• C(126°С, 3,35Па) |
|
|
• |
|
|
|
|
|
A(95.5°С, 0,496Па) |
|
|
|
E |
|
V |
|
Рис. 3. 7. Диаграмма состоя- |
|
|
|
T |
|
|
|
|
ния серы |
|
Превращение кристаллических структур может быть обратимым (энантиотропным) или необратимым (монотропным).
Классическим примером диаграммы состояния с энантиотропным полиморфным превращением является диаграмма серы (см. рис. 3. 7).
Точки А, В, С - точки трехфазового нонвариантного равновесия. Точка О - трехфазового метастабильного равновесия перегретых кристаллов ромбической серы, переохлажденной жидкой серы и пара, пересыщенного относительно равновесного пара моноклинной серы.
Если исходить из переохлажденной жидкой серы, то в первую оче-
40