


Тема9.СМЕЩЕНИЕ ХИМИЧЕСКОГО РАВНОВЕСИЯ
Под смещением химического равновесия понимают изменение концентраций веществ в реакционной смеси. Если при некотором воздействии в системе увеличиваются концентрации продуктов, то говорят, что равновесие смещается вправо, если увеличиваются концентрации реагентов, то равновесие смещается влево. Химическое равновесие можно сместить с помощью таких внешних факторов, как химическое воздействие (добавление или отбор тех или иных веществ), механическое воздействие (изменение давления, действующего на систему), тепловое воздействие (подвод или отвод теплоты).
Чтобы предсказать отклик равновесной системы на внешнее воздействие и определить направление смещения равновесия, используют принцип Ле Шателье (1884): если на систему, находящуюся в состоянии равновесия, оказывается внешнее воздействие, то равновесие сместится в направлении того процесса, протекание которого ослабляет эффект произведенного воздействия (в системе возникает самопроизвольныйпроцесс, компенсирующийданноевоздействие).
Принцип Ле Шателье следует из закона действующих масс. Если система находится при постоянной температуре, то константа равновесия при внешних воздействиях остается постоянной. Поэтому любое изменение равновесных концентраций (парциальных давлений) одного или нескольких веществ должно приводить к такому изменению равновесных концентраций (парциальных давлений) других веществ, чтобы соблюдалось постоянство константы равновесия.
В качестве примера рассмотрим реакцию
2(SO2) + (O2) 2(SO3); H298° = –198,4 кДж. (9.1)
Константа равновесия указанного процесса
KС = |
С2 (SO3 ) |
|
С2 (SO2 ) C(O2 ) . |
(9.2) |
1. Влияние концентраций (парциальных давлений) компонентов системы. При добавлении в реакционную смесь (9.1), например, кислорода, т. е. при увеличении С(O2), знаменатель в выражении для KС увеличивается, но поскольку KС – константа, величина постоянная при неизменной температуре, то должен увеличиться и числитель. Таким образом, в реакционной смеси возрастает количество продукта
85
реакции. Мы говорим о смещении химического равновесия вправо, в сторону продукта. Процесс будет протекать, пока не установится новое равновесие. Новые равновесные концентрации всех компонентов будут такими, чтобы соотношение между ними, определяемое константой равновесия (9.2), оставалось постоянным.
Если в систему добавить продукт реакции (увеличить концентрацию SO3), то в соответствии с принципом Ле Шателье равновесие сместится влево. Если концентрацию SO3 уменьшить, равновесие сместится вправо.
2. Влияние общего давления. По той же причине смещается равновесие реакции при изменении давления. Снова обратимся к реакции окисления оксида серы(IV). Повысим давление, скажем, в два раза. При этом объем газовой смеси уменьшится в два раза. Это значит, что концентрации всех газообразных веществ возрастут в два раза. В этом случае числитель выражения для KС увеличится в четыре раза, а знаменатель в восемь раз, т. е. отношение нарушится. Для его восстановления должны возрасти концентрация оксида серы(VI) и уменьшиться концентрации оксида серы(IV) и кислорода. Равновесие сместится вправо.
Увеличение общего давления смещает равновесие в сторону процесса, идущего с уменьшением числа молей газообразных веществ. Уменьшение общего давления газов в смеси будет смещать равновесие в сторону реакции, идущей с увеличением числа молей газообразных веществ.
Равновесие реакции
[FeO] + (H2) [Fe] + (H2O)
от давления не зависит, так как количества молей газообразных исходных веществ (Н2) и продуктов реакции (H2O) одинаковы.
Равновесие реакции
[С] + (СО2) 2(СО)
при повышении давления смещается влево (концентрация твердой фазы [С] не меняется).
3. Влияние температуры. Повышение либо понижение температуры означает приобретение либо потерю системой энергии и, следовательно, должно изменять величину константы равновесия.
Запишемуравнение–RT lnKР |
≈ |
H ° |
– Т |
S° |
в следующемвиде: |
|
|
|
298 |
|
298 |
||
RT lnKР = – Н° + Т S° ; |
|
|||||
lnKР = – |
|
Н° |
+ |
S° |
; |
|
|
|
R |
|
|||
|
RT |
|
|
|||
86
|
− H |
|
S |
|
298 |
+ |
298 |
KР = 10 |
19,15 T |
|
19,15 , |
где 19,15 = R · 2,303 (2,303 – коэффициент перехода от натурального логарифма к десятичному). В показателе степени первое слагаемое сильно зависит от температуры, второе слагаемое не содержит температуру и является постоянной величиной для процесса. Поэтому на-
правление смещения равновесия определяется знаком H298° .
Экзотермические реакции: H° < 0. В этом случае, чем выше температура, тем больше уменьшается KР. Таким образом, повышение температуры уменьшает величину константы равновесия, т. е. сме-
щает равновесие влево.
Эндотермические реакции: H° > 0. В этом случае повышение температуры увеличивает значение константы равновесия (смещает равновесие вправо).
Поскольку (9.1) – экзотермическая реакция, то с увеличением температуры равновесие смещается в сторону реакции, протекание которой приводит к поглощению теплоты, т. е. влево.
87
Тема10. ХИМИЧЕСКАЯ КИНЕТИКА.
СКОРОСТЬ ХИМИЧЕСКОЙ РЕАКЦИИ
Методы и приемы химической термодинамики позволяют выяснить лишь принципиальную возможность осуществления химической реакции, условия ее вероятного протекания, определить полноту превращения реагирующих веществ в продукты реакции. Законы химической термодинамики позволяют определить направление и предел протекания при данных условиях химического процесса, а также его термодинамические характеристики. Однако термодинамика не может ответить на вопросы о том, как осуществляется данный процесс и с какой скоростью. Эти вопросы – механизм и скорость химической реакции – и являются предметом химической кинетики.
Скорость химической реакции есть число ее элементарных актов, происходящих в единицу времени в единице объема (для гомогенных реакций) или на единице поверхности (для гетерогенных реакций).
Скорость химической реакции – изменение концентрации реа-
гирующих веществ ( С) в единицу времени (t).
Первое определение является наиболее строгим; из него следует, что скорость химической реакции можно также выражать как изменение во времени любого параметра состояния системы, зависящего от числа частиц какого-либо реагирующего вещества, отнесенное к единице объема или поверхности – электропроводности, оптической плотности, диэлектрической проницаемости и т. п. Однако наиболее часто в химии рассматривается зависимость концентрации реагентов от времени. Очевидно, что в случае односторонних (необратимых) химических реакций концентрации исходных веществ во времени постоянно уменьшаются ( С(исх. в-в) < 0), а концентрации продуктов реакции увеличиваются ( С(прод.) > 0). Скорость реакции считается положительной, поэтому математически определение средней скорости реакции в интервале времени t записывается следующим образом:
υ = ± |
С |
|
(10.1) |
|
t . |
||||
|
||||
Единицей измерения скорости является моль/(дм3 с). В различных интервалах времени средняя скорость химической реакции имеет разные значения; истинная (мгновенная) скорость реакции определяется как производная от концентрации по времени:
88

υист = ± |
dС |
. |
(10.2) |
|
|||
|
dt |
|
|
Для нахождения скорости реакции на основании экспериментальных данных строятся так называемые кинетические кривые – кривые зависимости концентрации участвующих в реакции веществ от вре-
мени (рис. 10.1):
C 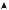
B
A
t
Рис. 10.1. Кинетические кривые для исходных веществ (А) и продуктов реакции (В)
Крутизна кинетической кривой в каждый момент времени характеризует истинную скорость реакции в этот момент времени, так как истинная скорость реакции в данный момент времени равна по абсолютной величине тангенсу угла наклона касательной к кинетической кривой (рис. 10.2):
υист = – ddtС = tgα.
C
α
t
Рис. 10.2. Графическое определение υист по зависимости С = f(t) исходного вещества
89
Если стехиометрические коэффициенты в уравнении химической реакции неодинаковы, величина скорости реакции будет зависеть от того, по изменению концентрации какого реагента она определялась. Очевидно, что в реакции
2Н2 + О2 = 2Н2О
имеет место следующее соотношение скоростей:
υ(Н2) = 2υ(О2) = υ(Н2О).
Молекулярность элементарных реакций
Элементарными (простыми) называют реакции, идущие в одну стадию. Их принято классифицировать по молекулярности. Молеку-
лярность элементарной реакции – число частиц, которые, согласно экспериментально установленному механизму реакции, участвуют в элементарном акте химического взаимодействия.
Мономолекулярные – реакции, в которых происходит химическое превращение одной молекулы (изомеризация, диссоциация и т. д.):
I2 = I• + I•.
Бимолекулярные – реакции, элементарный акт которых осуществляется при столкновении двух частиц (одинаковых или различных):
СН3Вr + KОН = СН3ОН + KВr.
Тримолекулярные – реакции, элементарный акт которых осуществляется при столкновении трех частиц:
О2 + NО + NО = 2NО2.
Реакции с молекулярностью более трех неизвестны.
Одной из задач, стоящих перед химической кинетикой, является определение состава реакционной смеси (т. е. концентраций всех реагентов) в любой момент времени, для чего необходимо знать зависи-
мость скорости реакции от концентраций. В общем случае, чем больше концентрации реагирующих веществ, тем больше скорость химической реакции. В основе химической кинетики лежит постулат химической кинетики: скорость химической реакции прямо пропорциональна произведению концентраций реагирующих веществ, взятых в некоторых степенях. Для реакции
аА + bВ + dD + ... = продукты реакции можно записать:
υ = k САх СВy СDz … |
(10.3) |
90
Коэффициент пропорциональности k есть константа скорости химической реакции. Константа скорости численно равна скорости реакции при концентрациях всех реагирующих веществ, равных 1 моль/л. Константа скорости зависит от тех же факторов, что и скорость, но не зависит отконцентрацииреагирующихвеществ.
Зависимость скорости реакции от концентраций реагирующих веществ определяется экспериментально и называется кинетическим уравнением химической реакции. Очевидно, что для того, чтобы запи-
сать кинетическое уравнение, необходимо экспериментально определить величину константы скорости и показателей степени при концентрациях реагирующих веществ. Показатель степени при концентрации каждого из реагирующих веществ в кинетическом уравнении химической реакции (в уравнении (10.3) соответственно x, y и z) есть частный порядок реакции по данному компоненту. Сумма показателей степени в кинетическом уравнении химической реакции (x + y + z) представляет собой общий порядок реакции. Следует подчеркнуть, что порядок реакции определяется из экспериментальных данных и не связан со стехиометрическими коэффициентами при реагентах в уравнении реакции. Для элементарных реакций порядок и молекулярность совпадают. Для сложных реакций эти понятия чаще всего не совпадают.
Для простых реакций показатели степеней в кинетических уравнениях равны коэффициентам стехиометрических уравнений. Например, скорость реакции H2 + I2 = 2HI может быть записана следующим образом:
υ = k · С(H2) С(I2),
где порядок реакции по водороду и иоду равен единице, а порядок реакции в целом равен 1 + 1 = 2. В этом случае стехиометрическое уравнение правильно отображает элементарный акт реакции.
Эта закономерность находит свое отражение в законе действующих масс, сформулированном норвежскими учеными К. Гульдбергом и П. Вааге (1867 г.): скорость простой реакции в каждый момент времени пропорциональна произведению концентраций реагирующих веществ, возведенных в степени, равные коэффициентам в стехиометрическом уравнении.
Зависимость скорости химической реакции от температуры.
Скорость химической реакции значительно зависит от температуры. Не всякое столкновение реагирующих частиц приводит к их взаимодействию. В химическое взаимодействие вступают только активные молекулы, т. е. обладающие энергией, достаточной для осуществления данной реакции. При повышении температуры число активных
91

молекул возрастает, так как нагревание сообщает молекулам необходимую энергию активации, т. е. ту дополнительную энергию, которая приводит к ослаблению химических связей в молекулах реагирующих веществ, а затем и к их разрыву.
Зависимость скорости химической реакции от температуры определяется эмпирическим правилом Вант-Гоффа: при повышении температуры на каждые 10 градусов скорость реакции возрастает примерно в 2–4 раза.
υ |
= υ γ |
T2 − T1 |
(10.4) |
10 |
|||
T |
T |
|
|
2 |
1 |
|
|
где υT2 и υT1 – скорость реакции при температуре Т2 и Т1 соответствен-
но, Т2 > Т1; γ – температурный коэффициент скорости реакции, показывающий, во сколько раз увеличивается скорость реакции при повышении температуры на каждые 10 градусов. Для большинства реакций при обычных температурах значения γ находятся в пределах 2–4.
Пример 1. На сколько градусов следует повысить температуру системы, чтобы скорость протекающей в ней реакции возросла в 50 раз? Температурный коэффициент скорости реакции равен 1,8.
Решение. Найдем значение Т = Т2 – Т1, при котором отношение
υT2 = 50.
υT1
Согласно правилу Вант-Гоффа |
|
υT2 |
= γ |
T2 − T1 |
||
|
10 |
, отсюда |
||||
|
|
|||||
|
|
υ |
|
|
|
|
|
|
T |
|
|
|
|
|
|
|
1 |
|
|
|
Т |
|
Т lg1,8 = lg50. |
||||
1,8 10 = 50 или |
|
|||||
|
10 |
|
|
|
|
|
Следовательно, Т = 67 град.
Правило Вант-Гоффа имеет ограниченное применение и предназначено для приблизительной оценки влияния температуры на скорость реакции. Более точно температурную зависимость скорости химической реакции отражает уравнение Аррениуса.
Уравнение Аррениуса
Очевидно, что взаимодействие частиц осуществляется при их столкновениях; однако число столкновений молекул очень велико, и, если бы каждое столкновение приводило к химическому взаимодействию частиц, все реакции протекали бы практически мгновенно.
92

С. Аррениус постулировал, что столкновения молекул будут эффективны (т. е. будут приводить к реакции) только в том случае, если сталкивающиеся молекулы обладают некоторым запасом энергии – энергией активации.
Энергия активации – минимальная избыточная энергия, которой должны обладать молекулы, по сравнению со средней энергией молекул, чтобы их столкновение могло привести к химическому взаимодействию.
Рассмотрим путь некоторой элементарной реакции: А2 + В2 → 2А—В.
Поскольку химическое взаимодействие частиц связано с разрывом старых химических связей и образованием новых, считается, что всякая элементарная реакция проходит через образование некоторого неустойчивого промежуточного соединения (переходного состояния), называемого активированным комплексом:
A |
|
B |
|
A |
|
|
|
|
|
|
|
|
|
|
B |
|
A |
|
|
|
B |
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
A |
|
B |
|
A |
|
|
|
|
|
|
|
|
|
|
|
B |
|
A |
|
|
|
B |
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
Переходное состояние – состояние системы, при котором уравновешены разрушение и создание связи: старые связи еще не разорваны, но уже ослаблены, новые связи наметились, но еще не образовались. В переходном состоянии система находится в течение небольшого времени (порядка 10−13 с). Энергия, которую необходимо затратить, чтобы привести систему в переходное состояние, и есть энергия активации. Образование активированного комплекса всегда требует затраты некоторого количества энергии, что вызвано, во-первых, отталкиванием электронных оболочек и атомных ядер при сближении частиц и, во-вторых, необходимостью построения определенной пространственной конфигурации атомов в активированном комплексе и перераспределения электронной плотности. Чем более жесткими являются требования ориентации реагентов при химическом взаимодействии, тем медленнее проходит химическая реакция. Реакции, характеризующиеся высокими значениями энергии активации, при низких температурах идут с малыми скоростями.
На рис. 10.3 приведены энергетические диаграммы экзотермической (а) и эндотермической (б) реакций, показывающие соотношение между энергией активации (E'а) и величиной теплового эф-
фекта ( Н).
93

путь реакции |
путь реакции |
а |
б |
Рис. 10.3. Энергетические диаграммы для экзотермической (а) и эндотермической (б) реакций
Очевидно, что если прямая реакция является экзотермической, то энергия активации обратной реакции (E''а) выше, чем энергия активации прямой реакции (E'а).
Экзотермические реакции протекают с меньшей энергией активации, чем эндотермические (рис. 10.3). Высокая энергия активации является причиной того, что многие термодинамически возможные химические реакции( G < 0) принизкихтемпературахнепротекают.
Если энергия столкновения молекул больше или равна энергии активации, то энергетический барьер преодолевается, и происходит образование продукта. В противном случае имеет место упругое столкновение молекулисходныхвеществ.
Поскольку температура есть мера средней кинетической энергии частиц, повышение температуры приводит к увеличению доли частиц, энергия которых равна или больше энергии активации, что приводит к увеличению константы скорости реакции.
Зависимость константы скорости реакции от температуры и величины энергии активации описывается уравнением Аррениуса:
Ea |
, |
(10.5) |
k = A е− RT |
где А – предэкспоненциальный множитель, отражающий число столкновений и ориентацию реагирующих частиц. Принципиально возможная реакция протекает при соблюдении двух условий: достаточной энергии и надлежащей ориентации частиц, благоприятствующей перераспределению электронной плотности; е – основание натурального логарифма (е = 2,718); Еа – энергия активации, Дж · моль–1; R – универсальная газоваяпостоянная(8,314 Дж· моль–1 · К–1); Т– температура, К.
94
Следовательно, увеличение скорости реакции при повышении температуры обусловлено главным образом резким увеличением доли активных соударений.
Как следует из уравнения Аррениуса, константа скорости реакции тем больше, чем меньше энергия активации. Зная энергию активации реакции и константу скорости при какой-либо температуре T1, по уравнению Аррениуса можно рассчитать величину константы скорости при любой температуре T2. Уравнение (10.5) позволяет также рассчитать энергию активации процесса по известным константам скорости при разных температурах.
Пример 2. Чему равна энергия активации реакции, если при повышении температуры от 290 до 300 К скорость ее увеличится в 2 раза?
Решение. Обозначим константы скорости реакции при 290 и 300 Ксоответственноk1 и k2. ИспользуяуравнениеАррениусанаходим:
|
k2 = e |
− Ea |
|
|
|
|
− Ea |
1 |
|
1 |
|
||||
|
300R |
|
|
|
− |
||||||||||
|
|
= e R |
|
300 |
290 |
|
|||||||||
|
|
|
|
|
|
. |
|||||||||
|
k1 |
|
|
|
− Ea |
|
|
|
|
|
|
|
|
|
|
|
|
e290R |
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
||||
Известно, что |
k 2 = 2, отсюда |
|
|
|
|
|
|
|
|||||||
|
k1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
− Ea |
|
1 |
− |
1 |
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
= 2. |
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
e R |
300 |
|
290 |
|
|
|
|||||||
Выражаем из последнего соотношения энергию активации:
|
ln2 = 2,303 lg2 = – |
Еа |
|
1 |
− |
1 |
|
; |
||
|
|
|
|
|
|
|||||
|
8,314 |
300 |
290 |
|||||||
|
0,693 8,314 |
|
|
|
|
|
||||
Еа = |
= 48 013 Дж/моль = 48,0 кДж/моль. |
|||||||||
0,003 45 − 0,003 33 |
||||||||||
Кинетика гетерогенных процессов
Гетерогенными называются процессы, происходящие на поверхности раздела соприкасающихся фаз. К таким процессам можно отнести реакции горения топлива, окисления металлов кислородом воздуха, процессы разложения вещества при электролизе, а также многие физические процессы: растворение газов и твердых тел в жидкостях, кристаллизация чистых жидкостей и растворов и др.
В системах из одного компонента гетерогенные процессы сводятся к переходу его из одной фазы в другую без изменения химического состава фаз. Сюда относятся процессы плавления, испарения, возгон-
95
ки и противоположные процессы кристаллизации и конденсации. Все эти процессы взаимно обратимы.
В системах из двух и более компонентов взаимодействие на поверхности раздела фаз приводит к возникновению различия в составах поверхностного и внутреннего слоев данной фазы.
Скорость гетерогенных процессов зависит от размеров и состояния поверхности раздела фаз. Гетерогенные процессы протекают, как правило, в несколько стадий, среди которых можно выделить стадию подвода к поверхности исходных веществ, стадию основного процесса, протекающего на поверхности раздела фаз, стадию отвода продуктов реакции. Так как эти стадии протекают последовательно, скорость суммарного процесса определяется наиболее медленной стадией.
Катализ Катализ – явление изменения скорости химической реакции в
присутствии особых веществ – катализаторов. Катализаторы – вещества, которые изменяют скорость химической реакции вследствие многократного участия в промежуточном химическом взаимодействии с реагентами, но после каждого цикла промежуточного взаимодействия восстанавливают свой химический состав.
Катализаторы одинаково ускоряют как прямую, так и обратную реакцию, и поэтому не смещают химическое равновесие. Они способствуют более быстрому достижению равновесного состояния.
Положительные катализаторы ускоряют реакцию, отрицательные (ингибиторы) замедляют реакцию. Ускоряющее действие катализатора заключается в уменьшении энергии активации катализируемой реакции. Например, для некаталитического разложения NH3 на N2 и Н2 Eа ≈ 320 кДж/моль, для того же разложения в присутствии Pt Eа ≈ 150 кДж/моль. Благодаря снижению Eа обеспечивается ускорение каталитических реакций по сравнению с некаталитическими.
Типы катализа: гомогенный, гетерогенный, автокатализ. Гомогенный катализ – каталитические реакции, в которых реа-
генты и катализатор находятся в одной фазе. В случае гомогеннокаталитических процессов катализатор образует с реагентами промежуточные реакционноспособные продукты, для чего требуется меньшее значение энергии активации.
Примером такого процесса может служить реакция разложения ацетальдегида, энергия активации которой Eа = 190 кДж/моль:
СН3СНО → СН4 + СО.
96
В присутствии паров иода этот процесс протекает в две стадии: СН3СНО + I2 → СН3I + НI + СО;
СН3I + НI → СН4 + I2.
Уменьшение энергии активации этой реакции в присутствии катализатора составляет 54 кДж/моль; константа скорости реакции при этом увеличивается приблизительно в 105 раз.
Наиболее распространенным типом гомогенного катализа является кислотный катализ, при котором в роли катализатора выступают ионы водорода Н+.
Гетерогенный катализ – каталитическая реакция, в которой катализатор и реагенты находятся в разных фазах и имеют границу раздела. На поверхности твердого катализатора (на поверхности раздела фаз) реагируют газообразные или жидкие вещества.
Механизм каталитических гетерогенных реакций очень сложен и зависит от природы реакции. Вначале на активных центрах поверхности твердого катализатора происходит процесс хемосорбции молекул исходных веществ. В результате происходит: a) локальное увеличение концентрации молекул исходных веществ на активных центрах; б) ослабление химических связей в молекулах исходных веществ, что заставляет их распадаться до атомов, образующиеся атомы реагируют друг с другом и образуют молекулу нового вещества, покидающую поверхность катализатора. К каталитическим гетерогенным реакциям относится реакция получения аммиака (катализатор – металлическое железо с примесью оксидов алюминия и калия):
3H2 + N2 2NH3.
Примером гетерогенного катализа является окисление SO2 в SO3 на катализаторе V2O5 при производстве серной кислоты (контактный метод):
2SO2 + O2 2SO3.
Автокатализ – процесс каталитического ускорения химической реакции одним из ее продуктов. В качестве примера можно привести катализируемую ионами водорода реакцию гидролиза сложных эфиров. Образующаяся при гидролизе кислота диссоциирует с образованием протонов, которые ускоряют реакцию гидролиза. Особенность автокаталитической реакции состоит в том, что данная реакция протекает с постоянным возрастанием концентрации катализатора. Поэтому в начальный период реакции скорость ее возрастает, а на последующих стадиях в результате убыликонцентрацииреагентов онаначинаетуменьшаться.
97
Тема11. ОСНОВНЫЕ ХАРАКТЕРИСТИКИ
РАСТВОРОВ
При смешении двух и более компонентов может образоваться механическая смесь или раствор (при условии отсутствия химического взаимодействия).
Механическая смесь – это система, где компоненты никоим образом не взаимодействуют друг с другом. Растворам присуще взаимодействие между компонентами (происходят процессы диссоциации, сольватации и др.). Любой раствор отличается от химического соединения тем, что его состав может в известных пределах меняться.
Растворы подразделяют на истинные и коллоидные. Если два компонента полностью растворяются друг в друге, образуя гомогенную (однофазную) систему, получается истинный раствор.
Истинный раствор – это гомогенная (однофазная) термодинамически устойчивая система, состоящая из двух или более компонентов, состав которой можно непрерывно изменять в некоторых пределах без скачкообразного изменения ее свойств.
В истинных растворах каждый компонент распределен в массе другого в виде молекул, атомов или ионов, например раствор хлорида натрия в воде является гомогенной системой из молекул воды, между которыми распределены ионы Na+ и Cl–.
Если компоненты нерастворимы или ограниченно растворимы друг в друге, то образуется гетерогенная система. Если в этой гетерогенной системе одна из фаз раздроблена до частиц мельчайших размеров (10–7–10–9 м), то получается коллоидный раствор.
Коллоидный раствор – это гетерогенная система, состоящая из двух или более фаз, причем одна из фаз (дисперсная) раздроблена до частиц или капель с размерами 10–7–10–9 м и распределена в другой сплошной фазе (дисперсионной среде). Коллоидный раствор, как правило, термодинамически неустойчивая система.
Растворы классифицируют в зависимости от агрегатного состояния на газообразные (воздух), жидкие, твердые (сплавы, стекла).
Компонент, агрегатное состояние которого при образовании раствора не изменяется, принято считать растворителем, а другой компонент растворенным веществом. Относительное содержание компонентов в растворе может быть любым. Оно ограничено лишь взаимной растворимостью веществ, которая зависит от их химической природы, сродства друг к другу, а также условий приготовления растворов
98
(температуры, давления для растворенных газов), присутствия других растворенных веществ.
Раствор, в котором не достигнут предел растворимости вещества, называется ненасыщенным раствором.
Раствор, в котором достигнут предел растворимости вещества, называется насыщенным раствором этого вещества. Растворимость вещества в том или ином растворителе характеризуется составом его насыщенного раствора. Наиболее распространенным способом характеристики состава насыщенного раствора служит коэффициент растворимости вещества. Коэффициент растворимости вещества – наибольшая масса вещества, способная при данной температуре раствориться в 100 г растворителя. Например, при 20оС в 100 г H2O с образованием насыщенного раствора растворяется 36 г NaCl (коэффициент растворимости равен 36 г на 100 г воды (20оС)).
Если в 100 г воды растворяется более 1,0 г вещества, то такое вещество называют хорошо растворимым. Если растворяется 0,1–1,0 г вещества – вещество малорастворимо. Наконец, вещество считают практически нерастворимым, если в 100 г воды растворяется менее 0,1 г вещества. Абсолютно нерастворимых веществ не бывает. Смотрите таблицу растворимости:
P – хорошо растворимые (более 1,0 г на 100 г воды); M – малорастворимые (0,1–1,0 г на 100 г воды);
Н – нерастворимые (менее 0,1 г на 100 г воды).
Существует еще один вид растворов – это пересыщенные растворы. Если насыщенный раствор, полученный при повышенной температуре, слить с кристаллов и дать ему охладиться, то получится такая жидкость, в которой больше растворенного вещества, чем это полагалось бы по значению его растворимости. Такой раствор – пересыщенный. Пересыщенные растворы очень неустойчивы. Помешивание, встряхивание, добавление крупинок соли может вызвать кристаллизацию избытка соли и переход в насыщенное устойчивое состояние.
Если содержание растворенного вещества в растворе (неважно, насыщенном или ненасыщенном) сравнительно маленькое, то раствор считается разбавленным, если большое – концентрированным.
Разбавленные растворы – это растворы, в которых массовая доля растворенного вещества составляет всего несколько процентов или молярная концентрация меньше 0,1 моль/л.
Вконцентрированных растворах массы растворенного вещества
ирастворителя сопоставимы.
99
Растворимость большинства (но не всех) твердых веществ с увеличением температуры увеличивается, а растворимость газов, наоборот, уменьшается. Это связано, прежде всего, с тем, что молекулы газов при тепловом движении способны покидать раствор гораздо легче, чем молекулы твердых веществ.
Измерение растворимости веществ при разных температурах показывает, что одни вещества заметно меняют свою растворимость в зависимости от температуры, другие – не очень сильно.
Если полученные в опытах значения нанести на оси координат, то получаются так называемые кривые растворимости различных веществ (рис. 11.1).
Растворимость
1
|
3 |
2 |
4 |
|
T
Рис. 11.1. Кривые растворимости некоторых солей в воде:
1 – KNО3; 2 – Nа2SО4 · 10Н2О; 3 – Nа2SО4; 4 – Ва(NО3)2
Эти кривые имеют практическое значение. По ним легко узнать, сколько вещества (например, KNO3) выпадет в осадок при охлаждении до 20°С насыщенного раствора, приготовленного при 80°С. С помощью таких операций очищают вещества. При охлаждении ненасыщенного раствора образуется насыщенный раствор, но насыщенный по основному веществу, которого больше всего, а не по примесям. По-
этому при охлаждении в осадок выпадает только чистое вещество, а примеси (вместе с частью вещества) остаются в растворе. Чистые кристаллы отфильтровывают от охлажденного, загрязненного примесями раствора. Этот способ очистки называется перекристаллизацией.
Давление не оказывает заметного влияния на растворимость твердых веществ, потому что при растворении не происходит заметного изменения объема системы. Зато увеличение давления повышает растворимость
100
газов. В этом можно убедиться, открыв бутылку с минеральной водой, в которой углекислый газ растворяют под давлением. Как только бутылку открывают, давление в ней падает и тут же уменьшается растворимость газа, которыйначинаетвыделяться израствораввидепузырьков.
Когда молекулы растворенного вещества связываются с молекулами воды, то получаются, строго говоря, новые химические соединения. Их общее название – гидраты, они, как правило, не имеют постоянного состава. Процесссвязываниявеществ сводойназывается гидратацией.
Поскольку образование водородных и других связей энергетически выгодно (для растворимых веществ), гидратация сопровождается выделением энергии. Часть этой энергии расходуется на разрушение кристаллической решетки, а ее избыток может выделяться в виде тепла. Например, растворение твердого гидроксида натрия NaOH сопровождается сильным разогревом раствора.
Если на разрушение кристаллической решетки тратится больше энергии, чем выделяется при получении гидратов, то раствор может охлаждаться. Например, если в стакан с водой поместить твердый нитрат аммония NH4NO3 и поставить стакан на влажный картон, то картон примерзает к стакану – настолько низко падает температура раствора. Растворение в водеNаNО3, KCl, K2SО4, NH4Cl – такжеэндотермическиепроцессы.
Молекулы воды из гидратной оболочки иногда могут вступать в химическую реакцию с растворенным веществом, образуя уже настоящее химическое соединение с постоянным составом, которые можно выделить из раствора, осторожно упаривая воду. Эти соединения называются кристаллогидратами, например CaSO4 2H2O.
При работе с растворами необходимо знать их количественный состав. Количественный состав растворов выражается различными способами. Наибольшее практическое применение в технике и лабораторной практике нашли следующие способы.
1. Массовая доля ω – число граммов растворенного вещества в 100 г раствора. Например, форма записи «раствор NaCl с массовой долей ω(NaCl) = 25%» означает, что 25 г NaCl содержатся в 100 г раствора. Масса воды в нем равна 75 г.
Массовая доля растворенного вещества ω(Х) – безразмерная величина, равная отношению массы вещества m(Х) к массе раствора m(р-ра):
ω(Х) = |
m(X ) |
, |
(11.1) |
|
m(р-ра) |
||||
|
|
|
где m(р-ра) = m(Х) + m(р-ля).
Массовую долю выражают в долях единицы или в процентах.
101

2. Молярная концентрация С(X) – число молей n растворенного вещества в одном литре раствора. Молярная концентрация равна отношению химического количества растворенного вещества (п(Х), моль) к объему раствора (V(р-ра), л):
С(Х) = |
n(X ) |
. |
(11.2) |
|
V (р-ра) |
||||
|
|
|
Химическое количество вещества выражается соотношением
n(Х) = |
m(X ) |
, |
(11.3) |
|
M (X ) |
||||
|
|
|
гдеm(Х) – массавеществаХ, г; М(Х) – молярная массавеществаХ, г/моль. Подставляя формулу (11.3) в (11.2), получаем
С(Х) = |
m(X ) |
, |
(11.4) |
M (X ) V (р-ра) |
Если известна масса т и плотность ρ раствора, то его объем
V(р-ра) = m(р-ра) . |
(11.5) |
ρ(р-ра) |
|
Молярная концентрация выражается в моль/л или моль/дм3 (эта размерность часто обозначается М). Например: 2 М NaOH или C(NaOH) = 2 моль/л. Данная форма записи означает, что 2 моля NaOH содержатся в 1 л раствора.
3. Молярная концентрация эквивалента (нормальная концен-
трация) С(1z X ) – число молей эквивалентов растворенного вещества nэкв в одном литре раствора.
Молярная концентрация эквивалента равна отношению химиче- |
||||||||||||||||
ского количества вещества эквивалента |
n( |
1 |
X ) |
или nэкв(X) |
к объему |
|||||||||||
z |
||||||||||||||||
раствора V(р-ра) (л): |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
C ( |
|
X ) = |
n |
( |
1 |
X ) |
|
|
|
|
m(X ) |
|
|
|
||
1 |
z |
= |
|
|
|
|
. |
(11.6) |
||||||||
z |
V |
(р-ра) |
M ( |
1 |
|
X ) V (р-ра) |
||||||||||
|
|
|
|
z |
|
|
|
|||||||||
Молярная концентрация эквивалента выражается в моль/л или моль/дм3 (часто обозначается н.). Например: 2 н. Na2SO4 или
С(12 Na2SO4 ) = 2 моль/л. Данная форма записи означает, что 2 моля эквивалента Na2SO4 содержатся в 1 л раствора.
102

Если взаимодействуют растворы веществ с известными нормальными концентрациями, то в точке эквивалентности справедливы соотношения:
n(1z X ) = n(1z Y );
С(1z X ) V(X) = С(1z Y ) V(Y).
4. Моляльная концентрация b – число молей растворенного вещества в одном килограмме растворителя.
b(X) = |
n(X ) |
= |
m(X ) |
, |
(11.7) |
|
m(р-ля) |
M (X ) m(р-ля) |
|||||
|
|
|
|
где n(X) – число молей вещества X; m – масса вещества Х; М – молярная масса вещества Х; m(р-ля) – масса растворителя, кг. Измеряют моляльность в моль/кг.
5. Молярная доля χ – это отношение количества компонента, содержащегося в данной системе (в молях), к общему количеству веществ в системе (в молях):
χ(Х) = |
n(X ) , |
(11.8) |
|
ni |
|
где n(Х) – количество вещества Х в растворе, моль; ni – количество вещества каждого компонента раствора, моль. Для двухкомпонентного раствора ni = n(Х) + n(S), где n(S) – количество вещества раство-
рителя S. Молярная доля также выражается в долях единицы или процентах. Сумма молярных (массовых) долей всех компонентов раствора равна 1.
Пример 1. Найти молярную концентрацию раствора с массовой долей 15% H2SO4 и плотностью 1,10 г/мл.
Решение. Пусть V(р-ра) = 1 л = 1000 мл. 1. Найдем массу 1 л раствора:
m(р-ра) = ρ(р-ра) V(р-ра) = 1,10 г/мл 1000 мл = 1100 г. 2. Найдем массу H2SO4 в 1 л раствора:
m(H2SO4) = ω(р-ра) m(р-ра) = 0,15 1100 = 165 г. 3. Найдем молярную концентрацию:
С(H2SO4 ) = |
m(H2SO4 ) |
= |
165 г |
= 1,68 моль/л. |
|||
M (H SO |
) V (р-ра) |
98 г/моль 1 л |
|||||
|
|
|
|||||
|
2 |
4 |
|
|
|
|
|
103

Пример 2. Сколько миллилитров 30%-ного раствора HCl (плотность 1,150 г/мл) понадобится для приготовления 1 л 0,1 н. раствора HCl?
Решение. 1) Найдем массу соляной кислоты, которая должна содержаться в приготовленном растворе:
m(HCl) = C(1z HCl) M(1z HCl) V(р-ра), где M(1z HCl) = 1 M(HCl) = 36,5 г/моль.
m(HCl) = 0,1 36,5 1 = 3,65 г.
2) Поскольку раствор готовят разбавлением водой 30%-ного раствора, то такая же масса HCl должна содержаться в 30%-ном растворе. Найдем массу 30%-ного раствора, содержащую 3,65 г HCl:
ω(HCl) = |
m(HCl) |
m(р-ра) = |
m(HCl) |
= |
3,65 |
= 12,167 г. |
|
m(р-ра) |
ω(HCl) |
0,3 |
|||||
|
|
|
|
3) Найдем объем 30%-ного раствора:
V(р-ра) = m(р-ра) = 12,167 = 10,58 мл. ρ(р-ра) 1,150
Пример 3. Сколько граммов Na2SO4 10H2O надо растворить в 800 г воды, чтобы получить раствор с массовой долей Na2SO4 10%?
Решение. Обозначим массу кристаллогидрата за х:
m(Na2SO4 10H2O) = х (г), тогда |
|
|
|
||
ω(Na2SO4) = |
m(Na2SO4 ) |
= |
m(Na2SO4 ) |
= 0,1. |
|
m(Na2SO4 10H2O) + m(H2O) |
|||||
|
|
x + 800 |
|
||
Выразим массу Na2SO4, содержащуюся в х г кристаллогидрата, используя молярные массы М(Na2SO4) = 142 г/моль, М(Na2SO4 10H2O) = = 142 + 180 = 322 г/моль.
322 г Na2SO4 10H2O содержат 142 г Na2SO4, х г Na2SO4 10H2O содержат m г Na2SO4:
m(Na2SO4) = 142322x .
Подставим m(Na2SO4) в выражение для ω и найдем х: |
||
ω(Na2SO4) = |
142 x |
= 0,1; |
322(x + 800) |
||
142 х = 0,1 322 (800 + х) = 25 760 + 32,2х; 109,8х = 25 760; х = 234,6.
104

m(Na2SO4 10H2O) = 234,6 г.
Пример 4. Сколько граммов СаСО3 выпадет в осадок, если к 800 мл 0,5 н. раствора СаСl2 прибавить избыток раствора Nа2СО3?
Решение. При сливании растворов СаСl2 и Nа2СО3 протекает реакция обмена: СаСl2 + Nа2СО3 = СаСО3↓ + 2NaCl.
Для решения задачи будем использовать закон эквивалентов: количества веществ эквивалентов реагирующих веществ и образующихся продуктов одинаковы, т. е.
n(1z CaCO3 ) = n(1z CaCl2 ) = 0,5 0,8 = 0,4 моль. m(CaCO3 ) = n(1z CaCO3 ) M (1z CaCO3 ) = 0,4 12 100 = 20 г.
105
Тема12. РАСТВОРЫ НЕЭЛЕКТРОЛИТОВ.
КОЛЛИГАТИВНЫЕ СВОЙСТВА РАСТВОРОВ
Вобщем случае при растворении происходит изменение свойств
ирастворителя, и растворенного вещества, что обусловлено взаимодействием молекул или ионов между собой по различным типам взаимодействия: Ван-дер-Ваальсового (во всех случаях), иондипольного (в растворах электролитов в полярных растворителях), специфических взаимодействий (образование водородных или донор- но-акцепторных связей). Учет всех этих взаимодействий представляет собой очень сложную задачу. Очевидно, что чем больше концентрация раствора, тем интенсивнее взаимодействие частиц, тем сложнее структура раствора. Поэтому количественная теория разработана только для идеальных растворов, единственной движущей силой об-
разования такого раствора является увеличение энтропии системы S; какие-либо тепловые или объемные эффекты при растворении отсутствуют ( Н = 0, V = 0). К идеальным можно отнести газовые растворы и растворы неполярных жидкостей, в которых энергия взаимодействия разнородных частиц E(A-B) близка к энергиям взаимодействия одинаковых частиц E(A-A) и E(B-B). Идеальными считают также бесконечно разбавленные растворы, в которых можно пренебречь взаимодействием частиц растворителя и растворенного вещества между собой. Свойства таких растворов зависят только от концентрации растворенного вещества, но не зависят от его природы.
Коллигативными свойствами раствора называются свойства,
зависящие от концентрации растворенного вещества и мало или совсем не зависящие от природы вещества. К таким свойствам относят:
−давление насыщенного пара растворителя;
−понижение температуры замерзания раствора;
−повышение температуры кипения раствора;
−осмотическое давление.
Давление насыщенного пара растворителя
Испарение – переход части молекул жидкости (воды) из жидкого агрегатного состояния в пар. Процесс испарения обратимый, одновременно происходит переход молекул из газовой фазы в жидкую (конденсация). С течением времени в закрытом сосуде с жидкостью при определенной температуре устанавливается равновесие, при ко-
106
тором скорость испарения равна скорости конденсации. Пар над жидкостью в состоянии равновесия считают насыщенным при данной температуре. Он производит определенное давление на поверхность жидкой фазы (давление насыщенного пара растворителя).
Допустим, в систему жидкость – пар, достигшую равновесия, внесено нелетучее вещество (например, хлорид натрия NaCl). Переход вещества в паровую фазу исключен. В растворе часть поверхности будет занята молекулами растворенного вещества. Кроме того, молекулы растворенного вещества и растворителя взаимодействуют между собой. Все это приводит к тому, что давление насыщенного пара растворителя над раствором уменьшается и зависит от количества растворенного вещества и природы растворителя.
Относительное понижение давления пара растворителя над раствором равно:
P0 |
− P |
= χ(в-ва), |
(12.1) |
|
P0 |
||||
|
|
|||
где Р0 – давление насыщенного пара растворителя над чистым растворителем; Р – давление насыщенного пара растворителя над раствором; χ(в-ва) – молярная доля растворенного вещества. Это уравнение является математической формулировкой закона Рауля (1886 г.): относительное понижение давления насыщенного пара над раствором равно молярной доле растворенного вещества и не зависит от природы вещества. Явление понижения давления насыщенного пара над раствором вытекает из принципа Ле Шателье.
Если в равновесную систему жидкость А пар введено некоторое вещество В, молярная доля растворителя χ(А) становится меньше
единицы; равновесие жидкость А пар в соответствии с принципом Ле Шателье смещается в сторону конденсации вещества А, т. е. в сторону уменьшения давления насыщенного пара Р(А). Очевидно, что чем меньше молярная доля растворителя А в растворе, тем меньше парциальное давление его насыщенных паров над раствором.
Давление пара идеальных и реальных растворов. Если компо-
ненты бинарного (состоящего из двух компонентов) раствора летучи, то пар над раствором будет содержать оба компонента (относительное содержание компонентов в парах будет, как правило, отличаться от содержания их в растворе – пар относительно богаче компонентом, температура кипения которого ниже). Рассмотрим бинарный раствор, состоящий из компонентов А и В, неограниченно растворимых друг в друге. Общее давление пара, согласно закону Рауля, равно:
107
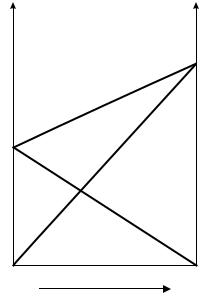
Р= Р0(А) · χ(А) + Р0(В) · χ(В) = Р0(В) · χ(В) + Р0(А) · (1 – χ(В)) =
=Р0(А) – χ(В) · (Р0(А) – Р0(В)).
Таким образом, для идеальных бинарных растворов зависимость общего и парциального давления насыщенного пара от состава раствора, выраженного в мольных долях компонента В, является линейной при любых концентрациях (рис. 12.1). К таким системам относятся, например, системы бензол – толуол, гексан – гептан, смеси изомерных углеводородов и др.
P |
P |
P0(B)
P0(A)

А |
В |
состав, χ(В)
Рис. 12.1. Зависимость парциальных и общего давлений пара идеального раствора от его состава
Для реальных растворов данные зависимости являются криволинейными. Если молекулы данного компонента взаимодействуют друг с другом сильнее, чем с молекулами другого компонента, то истинные парциальные давления паров над смесью будут больше (рис. 12.2, а), чем вычисленные по закону Рауля (положительные отклонения). Если же однородные частицы взаимодействуют друг с другом слабее, чем разнородные, парциальные давления паров компонентов будут меньше
(рис.12.2, б) вычисленных (отрицательные отклонения). Реальные растворы с положительными отклонениями давления пара образуются из чистых компонентов с поглощением теплоты ( Нраств > 0), растворы с отрицательными отклонениями образуются с выделением теплоты
( Нраств < 0).
108
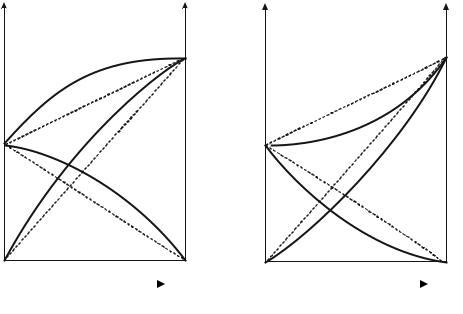
P |
P |
P |
P |
|
P0(B) |
|
P0(B) |
P0(A) |
P0(A) |
А |
В |
А |
В |
|
состав, χ(В) |
|
состав, χ(В) |
|
а |
|
б |
Рис. 12.2. Зависимость парциальных и общего давлений пара идеальных (штриховая линия) и реальных (сплошная линия) бинарных растворов от состава при положительных (а) и отрицательных (б) отклонениях от закона Рауля
Следствия из закона Рауля:
1.Растворение нелетучего компонента в растворителе приводит к расширению температурной области существования жидкой фазы.
2.Понижение температуры замерзания и повышение температуры кипения прямо пропорциональны моляльной концентрации растворенного вещества.
Понижение температуры замерзания раствора – криоскопия
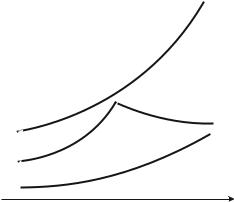
Из всех коллигативных свойств чаще других используются криоскопические измерения ввиду простоты и высокой точности измерения температуры замерзания. Последняя обычно измеряется с помощью специальных метастатических ртутных термометров (термометров Бекмана) с ценой деления 0,01 К. Такие термометры имеют шкалу только на 5 К, но снабжены дополнительным резервуаром для ртути, позволяющим настроитьегоналюбыеабсолютныетемпературыот260 до380 К.
Рассмотрим Р – T диаграмму состояния растворителя и растворов различной концентрации (рис. 12.3), на которой кривая ОF – зависимость давления насыщенного пара над твердым растворителем, а кривые ОА, ВС, DE – зависимости давления насыщенного пара над чистым растворителем и растворами с возрастающими концентрациями соответственно. Кристаллы растворителя будут находиться в равнове-
109

сии с раствором только тогда, когда давление насыщенного пара над кристаллами и над раствором одинаково. Поскольку давление пара растворителя над раствором всегда ниже, чем над чистым растворителем, температура, отвечающая этому условию, всегда будет более низкой, чем температура замерзания чистого растворителя. При этом понижение температуры замерзания раствора Tзам не зависит от природы растворенного вещества и определяется лишь соотношением числа частиц растворителя и растворенного вещества.
P A
C
O E
B
D
F
T2 T1 T0 зам |
T |
Рис. 12.3. Понижение температуры замерзания разбавленных растворов
Понижение температуры замерзания раствора Tзам прямо пропорционально моляльной концентрации вещества в растворе:
Tзам = K · b, |
(12.2) |
где Tзам – разность между температурой замерзания чистого растворителя (Т0 зам) и температурой начала кристаллизации (замерзания) раствора (Т1, Т2); K – криоскопическая константа, зависящая только от природы растворителя и не зависящая от природы растворенного вещества, физический смысл криоскопической постоянной – понижение температуры кристаллизации раствора с концентрацией вещества 1 моль/кг, для воды K = 1,86, для бензола K = 5,07; b – моляльная концетрация растворенного вещества, моль/кг.
Повышение температуры кипения раствора – эбуллиоскопия
Эбуллиоскопический метод основан на измерении температуры кипения растворов. Температура кипения растворов нелетучего вещества
110
всегда выше, чем температура кипения чистого растворителя при том же давлении. Рассмотрим Р – T диаграмму состояния растворителя и растворов различной концентрации (рис. 12.4). Любая жидкость – растворитель или раствор – кипит при той температуре, при которой давление насыщенного пара становится равным внешнему давлению. Соответственно температуры, при которых изобара Р = 1 атм пересечет кривые ОА, ВС и DE (рис. 12.3), представляющие собой зависимости давления пара над чистым растворителем и растворами с возрастающими концентрациями, будуттемпературамикипения этихжидкостей(рис. 12.4).
P |
A |
C |
E |
|
|
|
1 атм
O
B
D
T0 кип T1 T2
Рис. 12.4. Повышение температуры кипения разбавленных растворов
Повышение температуры кипения растворов нелетучих веществ
Tкип пропорционально понижению давления насыщенного пара и, следовательно, прямо пропорционально моляльной концентрации растворенного вещества:
Tкип = Е · b, |
(12.3) |
где Tкип – разность между температурой кипения чистого растворителя (Т0 кип) и температурой кипения раствора (Т1 и Т2); Е – эбуллиоскопическая константа, зависящая только от природы растворителя и не зависящая от природы растворенного вещества, физический смысл криоскопической постоянной – повышение температуры кипения раствора с концентрацией вещества 1 моль/кг, для воды Е = 0,52, для бензола Е = = 2,6; b – моляльная концентрациярастворенноговещества, моль/кг.
Рассмотренные методы позволяют определять давление насыщенного пара, температуры кипения, кристаллизации, молярные массы веществ.
111

Пример 1. Вычислить давление насыщенного пара раствора, содержащего 45 г глюкозы С6Н12О6 в 720 г воды при 25°С. Давление пара воды при 25°С составляет 23,76 мм рт. ст.
Решение. Р0 = 23,76 мм рт. ст., М(С6H12O6) = 180 г/моль, М(Н2О)= = 18 г/моль. Для решения задачи воспользуемся уравнением (12.1):
|
Р(H2O) = Р0 · χ(H2O) ; |
|
|||||
|
720 |
|
40 |
|
|
||
Р(H2O) = 23,76 |
18 |
|
= 23,76 |
|
= 23,61 мм рт. ст. |
||
720 + |
45 |
40,25 |
|
||||
|
18 |
|
180 |
|
|
|
|
Пример 2. Вычислить температуру кипения (Ткип) и температуру замерзания (Тзам) 4,6%-ного раствора глицерина С3Н8О3 в воде.
Решение. 100 г раствора содержат 4,6 г глицерина в 95,4 г воды,
М(С3Н8О3) = 92 г/моль, Е(H2O) = 0,52°, K(H2O) = 1,86°.
Для расчета используем приведенные выше формулы (12.2, 12.3):
Tзам = 1,86 1000 4,6 = 0,975°. 92 95,4
Tкип = 0,52 1000 4,6 = 0,27°; 92 95,4
Получаем искомые величины: Tзам = –0,975°С и Tкип = 100,27°С. 3. Растворы, содержащие одинаковое число молей растворенных веществ в одинаковом числе молей растворителя, обнаруживают одно и то же понижение температуры замерзания и одно и то же повыше-
ние температуры кипения.
Осмотическоедавлениераствора
Осмотическое давление связано с явлением осмоса. Осмос наблюдается в системах с полупроницаемыми перегородками, разделяющими раствор и растворитель или два раствора разной концентрации. Такие перегородки способны пропускать только молекулы растворителя. Число молекул растворителя, переходящих за единицу времени через полупроницаемую перегородку со стороны растворителя или болееразбавленного раствора, всегда больше числа тех молекул, которые проходят в обратном направлении. Со временем концентрации выравниваются, и система приходит в равновесное состояние. Такой самопроизвольный переход растворителя через полупроницаемую перегородку в раствор с большей концентрацией называют осмосом. Так как полупроницаемыми перегород-
112
ками являются оболочки растительных и животных клеток, то осмос служит одним из механизмов транспортировки растворителя – воды – вживотныхирастительныхорганизмах.
Мерой силы, с которой растворитель стремится перейти через полупроницаемую перегородку в раствор, является осмотическое давление, численно равное тому минимальному дополнительному давлению, которое нужно приложить к раствору, чтобы осмос прекратился со стороны чистого растворителя.
Растворы с одинаковым осмотическим давлением называются изотоническими. Если раствор по сравнению с другим имеет более высокое осмотическое давление, то его называют гипертоническим, а
с более низким – гипотоническим.
Осмотическое давление (Росм) прямо пропорционально молярной концентрации вещества в растворе (С) и абсолютной температуре (Т). Математически эта зависимость выражается уравнением Вант-Гоффа:
Росм = С · R · T,
где R – универсальная газовая постоянная.
Поскольку С = n/V, то формально уравнение Вант-Гоффа аналогично уравнению состояния идеального газа.
Измерение осмотического давления является одним из методов определения молекулярных масс, позволяющих в современных мембранных осмометрах определять Mr до 106 (каучук, целлюлоза, белки).
Вещество при изменении давления и температуры может переходить из одного агрегатного состояния в другое. Эти переходы, совершающиеся при постоянной температуре и давлении, называют фазовыми переходами первого рода.
Фаза – это совокупность гомогенных частей системы, имеющих одинаковый состав, химические и физические свойства и отделенных от других частей системы поверхностью раздела.
Количество теплоты, которое вещество получает из окружающей среды либо отдает ей при фазовом переходе, есть скрытая теплота фазового перехода. В гетерогенной системе, где нет химических взаимодействий, а возможны лишь фазовые переходы, при постоянстве температуры и давления существует так называемое фазовое равновесие. Фазовое равновесие характеризуется некоторым числом фаз, компонентов и числом степеней термодинамической свободы системы.
Компонент – химически однородная составная часть системы, которая может быть выделена из системы и существовать вне ее. Число независимых компонентов системы равно числу компонентов минус число возможных химических реакций между ними.
113
Число степеней свободы – число параметров состояния системы, которые могут быть одновременно произвольно изменены в некоторых пределах без изменения числа и природы фаз в системе.
Число степеней свободы гетерогенной термодинамической системы, находящейся в состоянии фазового равновесия, определяется правилом фаз, сформулированным Дж. Гиббсом: число степеней свободы равновесной термодинамической системы f равно числу независимых компонентов системы К минус число фаз Ф плюс число внешних факторов, влияющих на равновесие.
Для системы, на которую из внешних факторов влияют только температура и давление, можно записать:
f = К – Ф + 2.
Системы принято классифицировать по числу компонентов (од- но-, двухкомпонентные и т. д.), по числу фаз (одно-, двухфазные и т. д.) и числу степеней свободы (инвариантные, моно-, дивариантные и т. д.). Для систем с фазовыми переходами обычно рассматривают графическую зависимость состояния системы от внешних условий – так называемые диаграммы состояния.
Анализ диаграмм состояния позволяет определить число фаз в системе, границы их существования, характер взаимодействия компонентов. При непрерывном изменении параметров состояния все свойства отдельных фаз изменяются также непрерывно; свойства системы в целом изменяются непрерывно до тех пор, пока не изменится число или природа фаз в ней, что приводит к скачкообразному изменению свойств системы. На диаграмме состояния системы каждой фазе соответствует часть плоскости – поле фазы. Линии пересечения плоскостей отвечают равновесию между двумя фазами. Всякая точка на диаграмме состояния (т. н. фигуративная точка) отвечает некоторому состоянию системы с определенными значениями параметров состояния.
Рассмотрим диаграмму состояния воды (рис. 12.5). Поскольку вода – единственное присутствующее в системе вещество, число независимых компонентов К = 1. В системе возможны три фазовых равновесия: между жидкостью и газом (линия ОА – зависимость давления насыщенного пара воды от температуры), твердым телом и газом (линия ОВ – зависимость давления насыщенного пара надо льдом от температуры), твердым телом и жидкостью (линия ОС – зависимость температуры плавления льда от давления). Три кривые имеют точку пересечения О, называемую тройной точкой воды; тройная точка отвечает равновесию между тремя фазами.
114
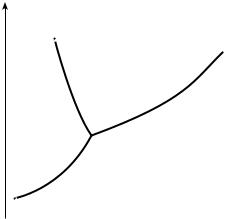
В тройной точке система трехфазна и число степеней свободы равно нулю; три фазы могут находиться в равновесии лишь при строго определенных значениях температуры и давления (для воды трой-
ная точка отвечает состоянию с Р = 0,61 кПа и Т = 0,01°С).
|
Кривая ОВ теоретически продол- |
P |
жается до абсолютного нуля, а кривая |
C |
давления насыщенного пара над жид- |
А |
костью ОА заканчивается в крити- |
ческой точке воды (Tкрит = 374°C и |
|
|
давлении 22,064 МПа (218 атм)); вы- |
|
ше критической температуры газ и |
Ожидкость не могут существовать как
отдельные фазы. Кривая ОС в верхней части (при высоких давлениях) изме-  няет свой наклон (появляются новые T кристаллические фазы, плотность которых, в отличие от обычного льда,
няет свой наклон (появляются новые T кристаллические фазы, плотность которых, в отличие от обычного льда,
выше, чем у воды).
Внутри каждой из областей диаграммы (АОВ, ВОС, АОС) система однофазна; число степеней свободы системы равно двум (система дивариантна), т. е. можно одновременно изменять и температуру, и давление, не вызывая изменения числа фаз в системе:
С = 1 – 1 + 2 = 2.
На каждой из линий число фаз в системе равно 2, и согласно правилу фаз система моновариантна, т. е. для каждого значения температуры имеется толькооднозначениедавления, прикоторомсистемадвухфазна:
С = 1 – 2 + 2 = 1.
Кривая равновесия ОС «твердое вещество – жидкость» на диаграммах состояния воды и висмута наклонена влево, а на диаграммах состояния остальных веществ – вправо. Это связано с тем, что плотность воды больше, чем плотность льда, т. е. плавление сопровождается уменьшением объема ( V < 0).
115