

Реакции моносахаридов в открытой форме
1. Окисление моносахаридов мягкими окислителями (гидроксид диаммминсеребра,гидроксид меди (II), бромноватистая кислота HBrO) дает альдоновые кислоты:

2. Восстановление амальгамой натрия, алюмогидридом лития или боргидридом натрия приводит к образованию шестиатомных спиртов:

При восстановлении альдогексоз иодоводородом при нагревании образуется 2-иодгексан.
3. Образование оксимов. Моносахариды легко реагируют с гидроксиламином NH2OH,дальнейшая дегидратация приводит к нитрилам, которые при отщеплении циановодорода образуют альдозы с меньшим числом атомов углерода. Так можно установить строение моносахарида и его принадлежность к D или L ряду.

Реакции моносахаридов в циклической форме
1. Образование
гликозидов. Гликозидный
гидроксил легко вступает в реакции со
спиртами,аминами, тиозами, образуя O, N
или S-гликозиды, например при действии
на ![]() -D-глюкопиранозу
этанола в присутствии соляной кислоты
образуется О-этилгликозид
-D-глюкопиранозу
этанола в присутствии соляной кислоты
образуется О-этилгликозид![]() -D-глюкопиранозы:
-D-глюкопиранозы:

Образующийся гликозид уже не способен к переходу в открытую форму.
2. Алкилирование происходит под действием алкилгалогенидов, при этом алкилируются все гидроксилы:

При
гидролизе образовавшейся
пентаэтил-![]() -D-глюкопиранозы
освобождается только гликозидный
гидроксил:
-D-глюкопиранозы
освобождается только гликозидный
гидроксил:

В
результате получается
тетрааэтил-![]() -D-глюкопираноза,
наличие свободного гликозидного
гидроксилапозволяет ей переходить в
открытую форму и, соответственно в
тетрааэтил-
-D-глюкопираноза,
наличие свободного гликозидного
гидроксилапозволяет ей переходить в
открытую форму и, соответственно в
тетрааэтил-![]() -D-глюкопиранозу:
-D-глюкопиранозу:

3. Ацилирование под
действием галогенангидридов или
ангидридов кислот приводитк образованию
ацильных производных, например при
ацетилировании ![]() -D-глюкопиранозы
образуется пентаацетил-
-D-глюкопиранозы
образуется пентаацетил-![]() -D-глюкопираноза:
-D-глюкопираноза:
Брожение моносахаридов
Брожение - расщепление молекул моносахаридов под действием ферментов (энзимов). Какправило, брожению подвергаются моносахариды с числом углеродных атомов, кратным трем. Моносахаридымогут подвергаться нескольким видамброжения. Наиболее известный - спиртовое брожение, протекает под действием фермента дрожжей зимазы:
С6Н12О6 = 2С2Н5ОН + 2СО2
При ацетонобутаноловом брожении образуются ацетон и бутанол, при лимоннокислом -лимонная кислота, маслянокислое брожение дает масляную (бутановую) кислоту.
21.1
КЛАЙЗЕНА КОНДЕНСАЦИЯ (ацилирование по
Клайзену, сложноэфирная конденсация),
взаимод. сложных
эфировкарбоновых
к-т в присут. основных катализаторов с
соед., содержащими активную метиленовую
группу, с образованием новой
углерод-углеродной связи:
 R
и R'-opr. радикалы, R: -
обычно COOAlk, C(O)Alk, CN. Для
осуществления процесса исходные в-ва
и катализатор(напр.,
Na, AlkONa, NaNH2,
NaH) кипятят в осушенном инертном р-рителе
(иногда до неск. сут). Для ускорения р-ции
в ряде случаев образующийся спирт отгоняют
в вакууме. При
самоконденсации сложных
эфиров образуются b-кетоэфиры.
Таким образом получают, напр., ацетоуксусный
эфир:
R
и R'-opr. радикалы, R: -
обычно COOAlk, C(O)Alk, CN. Для
осуществления процесса исходные в-ва
и катализатор(напр.,
Na, AlkONa, NaNH2,
NaH) кипятят в осушенном инертном р-рителе
(иногда до неск. сут). Для ускорения р-ции
в ряде случаев образующийся спирт отгоняют
в вакууме. При
самоконденсации сложных
эфиров образуются b-кетоэфиры.
Таким образом получают, напр., ацетоуксусный
эфир:
 Чтобы
избежать самоконденсации, р-цию проводят
в избытке ацилирующего эфира. Если
оба сложных
эфирасодержат
активные атомы Н,
образуется смесь четырех b-кетоэфиров,
что существенно снижает выход продуктов
перекрестной конденсации.
С помощью диэтилоксалата или этилформиата
можно также ацилировать этиловые
эфиры кротоновой
(n = 1) и сорбиновой (n = 2) к-т,
напр.:
НСООС2Н5+СН3(СН=СН)nСООС2Н5:НС(O)СН2(СН=СН)nСООС2Н5
Р-цией сложных
эфиров карбоновых
к-т с кетонами получают
р-дикетоны или b-кетоальдегиды:
Чтобы
избежать самоконденсации, р-цию проводят
в избытке ацилирующего эфира. Если
оба сложных
эфирасодержат
активные атомы Н,
образуется смесь четырех b-кетоэфиров,
что существенно снижает выход продуктов
перекрестной конденсации.
С помощью диэтилоксалата или этилформиата
можно также ацилировать этиловые
эфиры кротоновой
(n = 1) и сорбиновой (n = 2) к-т,
напр.:
НСООС2Н5+СН3(СН=СН)nСООС2Н5:НС(O)СН2(СН=СН)nСООС2Н5
Р-цией сложных
эфиров карбоновых
к-т с кетонами получают
р-дикетоны или b-кетоальдегиды:
 В
случае эфиров дикарбоновых к-т происходит
внутримол. конденсация (см. Дикмана
реакция). Клайзенаконденсация обратима
и по механизму близка к альдольной
конденсации,
напр.:
В
случае эфиров дикарбоновых к-т происходит
внутримол. конденсация (см. Дикмана
реакция). Клайзенаконденсация обратима
и по механизму близка к альдольной
конденсации,
напр.:

ДИКМАНА РЕАКЦИЯ (Дикмана конденсация),
внутримол. конденсация эфиров
двухосновных к-т в циклич. b-кетоэфиры,
напр.:
 Дикмана реакция -
частный случай Клайзена
конденсации (взаимод.
двух молекул сложного
эфира с
образованием ациклич. кетоэфира).
Осуществляется в присут. оснований (щелочные
металлы,
их гидрооксиды, алкоголяты,
амиды,гидриды,
реже - трифенилметилнатрий,
N-метиланилид лития и
др.) в инертной атмосфере в
среде эфира или ароматич. р-рителей. Как
правило, легко образуются 5- и 6-членные
циклы; в условиях большего разбавления
возможен синтез макроциклов. Присутствие
алкильных групп в a-
и b-положениях
к карбоксильной
группепрепятствует циклизации,
этоксикарбонильные группы в b-положении
облегчают. Из двух возможных продуктов
Дикмана реакции образуется
преимущественно продукт циклизации с
участием метиленовой группы, обладающей
более выраженными кислотными свойствами,
напр.:
Дикмана реакция -
частный случай Клайзена
конденсации (взаимод.
двух молекул сложного
эфира с
образованием ациклич. кетоэфира).
Осуществляется в присут. оснований (щелочные
металлы,
их гидрооксиды, алкоголяты,
амиды,гидриды,
реже - трифенилметилнатрий,
N-метиланилид лития и
др.) в инертной атмосфере в
среде эфира или ароматич. р-рителей. Как
правило, легко образуются 5- и 6-членные
циклы; в условиях большего разбавления
возможен синтез макроциклов. Присутствие
алкильных групп в a-
и b-положениях
к карбоксильной
группепрепятствует циклизации,
этоксикарбонильные группы в b-положении
облегчают. Из двух возможных продуктов
Дикмана реакции образуется
преимущественно продукт циклизации с
участием метиленовой группы, обладающей
более выраженными кислотными свойствами,
напр.:
 В
орг. синтезах часто используется р-ция,
обратная Дикмана реакции,
напр.:
В
орг. синтезах часто используется р-ция,
обратная Дикмана реакции,
напр.:
 Дикмана реакция применяется
для получения алициклич., полициклич.
и гетероциклич. соединений. Модификация
Дикмана реакции перегруппировка
N-замещенных фталимидов или изатинов в
производные изохинолина илихинолина (р-ция
Габриеля - Кольмана), напр.:
Дикмана реакция применяется
для получения алициклич., полициклич.
и гетероциклич. соединений. Модификация
Дикмана реакции перегруппировка
N-замещенных фталимидов или изатинов в
производные изохинолина илихинолина (р-ция
Габриеля - Кольмана), напр.:
 При
использовании в качестве основания Na
или NaOH наряду с продуктами Дикмана реакции в
больших кол-вах образуются
циклич. ацилоины. Р-ция
открыта В. Дикманом в 1894. О получении
макроциклич. кетонов циклизацийдинитрилов
см. Циглера
реакция. Лит..
Вульфсон Н. С., Зарецкий В. И., в кн.. Реакция и
методы исследованияорганических
соединений,
кн. 12, М., 1963, с. 7-257. Н.
Г. Гамбарян.
При
использовании в качестве основания Na
или NaOH наряду с продуктами Дикмана реакции в
больших кол-вах образуются
циклич. ацилоины. Р-ция
открыта В. Дикманом в 1894. О получении
макроциклич. кетонов циклизацийдинитрилов
см. Циглера
реакция. Лит..
Вульфсон Н. С., Зарецкий В. И., в кн.. Реакция и
методы исследованияорганических
соединений,
кн. 12, М., 1963, с. 7-257. Н.
Г. Гамбарян.
21.2
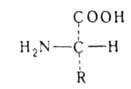
АМИНОКИСЛОТЫ, органические к-ты, содержащие одну или несколько аминогрупп. В зависимости от природы кислотной ф-ции аминокислоты подразделяют на аминокарбоновые, например H2N(CH2)5COOH, аминосульфоновые, например H2N(CH2)2SO3H, аминофосфоновые, например H2NCH[Р(О)(ОН)2]2, и аминоарсиновые, например H2NC6H4AsO3H2. Согласно правилам ИЮПАК, название
аминокислоты производят от названия соответствующей к-ты; взаимное расположение в углеродной цепи карбоксильной и аминной групп обозначают обычно цифрами, в нек-рых случаях - греч. буквами. Однако, как правило, пользуются тривиальными названиями аминокислот.
Структура и
физические свойства. По
физ. и ряду хим. свойств
аминокислоты резко
отличаются от соответствующих к-т
и оснований (см.
табл. 1 и 2). Они лучше раств. в воде,
чем в орг. р-рителях; хорошо кристаллизуются;
имеют высокую плотность и исключительно
высокие т-ры плавления (часто
разложения). Эти св-ва указывают на
взаимод. аминных и кислотных групп,
вследствие чего аминокислоты в твердом
состоянии и в р-ре (в широком интервале
рН) находятся в цвиттер-ионной форме.
Напр., для глицина кислотно-основное равновесие:

Взаимное влияние
групп особенно ярко проявляется
у![]() аминокислот,
где обе группы находятся в непосредств.
близости, а также у о- и n-аминобензойных
к-т, где их взаимод. передается через
систему сопряженных связей. Благодаря
электроноакцепторным св-вам группы
—
аминокислот,
где обе группы находятся в непосредств.
близости, а также у о- и n-аминобензойных
к-т, где их взаимод. передается через
систему сопряженных связей. Благодаря
электроноакцепторным св-вам группы
—![]() Н3 резко
усиливается кислотность карбоксильных
групп,
напр. рКа глицина 2,34,
тогда как уксусной к-ты
4,75,
Н3 резко
усиливается кислотность карбоксильных
групп,
напр. рКа глицина 2,34,
тогда как уксусной к-ты
4,75,![]() аланина 3,6. Аминогруппа подвергается
взаимокомпенсируемому влиянию
электроноакцепторной карбонильной
группы и электронодонорного отрицательно
заряженного атомакислорода,
в результате чего, напр.,
основность аминогрупп аминоуксусной
и n-аминобензойной к-т мало отличается
от основности
соотв. этиламина и анилина. Аминогруппа аминокислот
ионизирована в несколько меньшей
степени, чем карбоксильная
группа,
и водный р-р аминокислоты имеет слабокислый
характер. Значение рН, при
к-ром концентрация катионов аминокислоты
равна концентрации анионов,
наз. изоэлектрич. точкой (рI). Все
аминокислоты в изоэлектрич. точке имеют
минимум р-римости (в р-рах к-т
и щелочей р-римость
возрастает). Вблизи рI р-ры аминокислот
обладают миним. буферным действием, а
вблизи рК каждой функц. группы-максимальным.
аланина 3,6. Аминогруппа подвергается
взаимокомпенсируемому влиянию
электроноакцепторной карбонильной
группы и электронодонорного отрицательно
заряженного атомакислорода,
в результате чего, напр.,
основность аминогрупп аминоуксусной
и n-аминобензойной к-т мало отличается
от основности
соотв. этиламина и анилина. Аминогруппа аминокислот
ионизирована в несколько меньшей
степени, чем карбоксильная
группа,
и водный р-р аминокислоты имеет слабокислый
характер. Значение рН, при
к-ром концентрация катионов аминокислоты
равна концентрации анионов,
наз. изоэлектрич. точкой (рI). Все
аминокислоты в изоэлектрич. точке имеют
минимум р-римости (в р-рах к-т
и щелочей р-римость
возрастает). Вблизи рI р-ры аминокислот
обладают миним. буферным действием, а
вблизи рК каждой функц. группы-максимальным.
22.1
АЦИЛОИНОВАЯ КОНДЕНСАЦИЯ,
образование![]() гидроксикетонов (ацилоинов) восстановлением эфиров
алифатич. моно- или дикарбоновых к-т,
напр.:
гидроксикетонов (ацилоинов) восстановлением эфиров
алифатич. моно- или дикарбоновых к-т,
напр.:

Ацилоиновая конденсация эфиров
монокарбоновых к-т осуществляется двумя
путями:

Сложный эфир нагревают с Na (мольное соотношение 1:2) в бескислородной атмосфере в инертных р-рителях, напр. в бензоле, толуоле, ксилоле. Применение вместо Na жидкого сплава его с К позволяет проводить процесс при комнатной т-ре. Для повышения выхода ацилоинов в р-ции используют (CH3)3SiCl; образующиеся при этом бис-триметилсилиловые эфиры ендиолов легко выделяются и при взаимод. со спиртом образуют ацилоины. При осуществлении ацилоиновой конденсации без (CH3)3SiCl получаются побочные продукты-1,2-дикетоны, кетоны, а также вещества, синтезирующиеся в результате конденсации Дикмана и Клайзена.
С помощью ацилоиновой конденсации можно получать с хорошими выходами ациклич. ацилоины с R от СН3 до С22Н45. Р-ция с участием эфиров двух разных монокарбоновых к-т практически не осуществляется.
Из эфиров дикарбоновых
к-т в условиях, при к-рых происходит
ацилоиновая конденсация монокарбоновых
к-т, образуются циклич. ацилоины:

Таким путем можно
получить ацилоины,
содержащие в цикле от 4 до 42 атомов С.
Эту же реакцию используют
для синтеза макроциклических
гетероциклических ацилоинов и ацилоинов с
конденсированными циклами, напр.:

В результате
ацилоиновой конденсации эфира
дикарбоновой к-ты, содержащей 34 атома С,
в присут. 34-членногоциклоалкана удалось
получить катенан.
Ацилоиновая конденсация -
промежут. р-ция при получении
макроциклич.углеводородов,
макроциклич. и ациклич. кетонов,![]() дикетонов и
диолов.
дикетонов и
диолов.
22.2
Химические свойства. Р-ции по карбоксильным группам аминокислот, аминогруппа к-рых защищенаацилированием или солеобразованием, протекают аналогично превращениям карбоновых к-т. Аминокислоты легко образуют соли, сложные эфиры, амиды, гидразиды, азиды, тиоэфиры, галогенангидриды, смешанные ангидриды и т.д. Эфиры аминокислот под действием натрия или магнийорг. соед. превращаются в аминоспирты. При сухой перегонке в присут. Ва(ОН)2 аминокислоты декарбоксилируются.
Р-ции аминогрупп аминокислот
аналогичны превращениям аминов.
Аминокислоты образуют соли с
минер, к-тами и пикриновой к-той, легко
ацилируются хлорангидридами к-т
в водно-щелочном р-ре (р-ция Шоттена -
Баумана) и алкилируются алкилгалогенидами.
Метилиодид и диазометан превращают
аминокислоты в бетаины![]() .
С формалином аминокислоты
дают мегилольные или метиленовые
производные, а в присут. муравьиной к-ты
или каталитически активированного
Н2-N,N-диметиламинокислоты.
Под действием HNO2ароматич. аминогруппы диазотируются,
а алифатические замещаются на гидроксил.
При обработке эфиров
аминокислот изоцианатами и изотиоцианатами образуются
производные мочевины и тиомочевины.
При нагр. ссодой или
при одноврем. воздействии алкоголята и
СО2 аминокислоты
дают соли или
эфиры N-карбоксипроизводных аминокислот,
а при использовании CS2-аналогичные
дитиокарбаматы.
.
С формалином аминокислоты
дают мегилольные или метиленовые
производные, а в присут. муравьиной к-ты
или каталитически активированного
Н2-N,N-диметиламинокислоты.
Под действием HNO2ароматич. аминогруппы диазотируются,
а алифатические замещаются на гидроксил.
При обработке эфиров
аминокислот изоцианатами и изотиоцианатами образуются
производные мочевины и тиомочевины.
При нагр. ссодой или
при одноврем. воздействии алкоголята и
СО2 аминокислоты
дают соли или
эфиры N-карбоксипроизводных аминокислот,
а при использовании CS2-аналогичные
дитиокарбаматы.
Р-ции с одноврем.
участием групп NH2 и
СООН наиб. характерны для![]() .,
к-рые способны образовывать устойчивые
5-членные гетероциклы. С ионами переходных
металлов (Си,
Zn, Ni, Co, Pb, Ag, Hg, Cr)
.,
к-рые способны образовывать устойчивые
5-членные гетероциклы. С ионами переходных
металлов (Си,
Zn, Ni, Co, Pb, Ag, Hg, Cr)![]() .
образуют прочные хелатные комплексы,
что используется в комплексонах и
в комплексообразующих
ионообменных смолах на
основе аминокарбоновых и аминофосфоновых
к-т. При взаимод. с фосгеном
.
образуют прочные хелатные комплексы,
что используется в комплексонах и
в комплексообразующих
ионообменных смолах на
основе аминокарбоновых и аминофосфоновых
к-т. При взаимод. с фосгеном![]() .
превращаются в циклич.ангидриды N-карбоксиаминокислот
(ф-ла I), а при нагр. с уксусным
ангидридом или ацетилхлоридом -
в азлактоны (II); нагревание аминокислот
с мочевиной или
обработка изоцианатами дает гидантоины (III),
а при использовании
.
превращаются в циклич.ангидриды N-карбоксиаминокислот
(ф-ла I), а при нагр. с уксусным
ангидридом или ацетилхлоридом -
в азлактоны (II); нагревание аминокислот
с мочевиной или
обработка изоцианатами дает гидантоины (III),
а при использовании![]() .,
и особенно легко их эфиры, при нагр.
превращаются в 2,5-пиперазиндионы,
или дикетопиперазины (V).
.,
и особенно легко их эфиры, при нагр.
превращаются в 2,5-пиперазиндионы,
или дикетопиперазины (V).![]() .
при нагр. дезаминируются и
образуют
.
при нагр. дезаминируются и
образуют![]() -ненасыщенные
к-ты,
-ненасыщенные
к-ты,![]() и
и![]() .
отщепляют воду и
образуют 5- и 6-членные лактамы.
.
отщепляют воду и
образуют 5- и 6-членные лактамы.![]() Аминокапроновая
к-та при нагр. образует в осн. полиамид и
лишь частично превращ. вкапролактам,
что характерно и для аминокислот с
большим числом метиленовых звеньев
между функц. группами.Бетаины
Аминокапроновая
к-та при нагр. образует в осн. полиамид и
лишь частично превращ. вкапролактам,
что характерно и для аминокислот с
большим числом метиленовых звеньев
между функц. группами.Бетаины![]() .
при нагр. могут обратимо превращ. в эфиры
диметиламинокислот, напр.:
.
при нагр. могут обратимо превращ. в эфиры
диметиламинокислот, напр.:![]() .
При элиминировании триметиламина оетаины
.
При элиминировании триметиламина оетаины![]() .
превращ. в ненасыщ. к-ты,
.
превращ. в ненасыщ. к-ты,![]() и
и![]() -бетаины-в
циклич. лактоны.
При окислении
-бетаины-в
циклич. лактоны.
При окислении![]() .
образуют альдегиды с
укороченной углеродной цепочкой. Из-за
положит. заряда на четвертичном атоме N бетаины не
образуют солей сощелочами.
По аналогичной причине аминосульфоновые
и аминофосфоновые к-ты не образуют солей с
к-тами.арилизотиоцианатовтиогидантоины
(IV).
.
образуют альдегиды с
укороченной углеродной цепочкой. Из-за
положит. заряда на четвертичном атоме N бетаины не
образуют солей сощелочами.
По аналогичной причине аминосульфоновые
и аминофосфоновые к-ты не образуют солей с
к-тами.арилизотиоцианатовтиогидантоины
(IV).

23.1
АМИДЫ КАРБОНОВЫХ КИСЛОТ [от ам(миак)], ацилпроизводные аммиака или аминов, соед. общей ф-лы RC(O)NR'R". Незамещенные у атома N амиды RCONH2 наз. первичными; моно- и дизамещенные амиды RCONHR' и RCONR'R" (R' и R"-opr. остаток) - соотв. вторичными и третичными. Соед., содержащие две ацильные группы уатома азота RCON(R')COR", наз. имидами, а соединения с тремя ацильными группами RCON(COR')COR" - триациламинами. По др. классификации соед., содержащие одну, две или три ацильные группы, наз. соотв. первичными, вторичными или третичными. Циклич. аналогами амидов являются лактамы. Об амидах суль-фокислот см. Сульфамиды.
Названия первичных
амидов производят от названий
соответствующих к-т, напр. НСОNН2-формамид,
или амид муравьиной к-ты, СН3СОNH2-ацетамид,
или амид уксусной к-ты, С6Н5СОNН2 - бензамид,
или амид бензойной к-ты. В названиях
N-замещенных амидов заместители
перечисляются перед названием
незамещенного амида, напр. HCON(CH3)2 -
N.N-диметилформамид.

Кроме жидких формамида и N-метил-формамида, первичные и вторичные амиды-кристаллич. в-ва, большинство третичных-жидкости. Низшие алифатич. амиды хорошо раств. в воде, простейшие ароматические - умеренно в горячей воде. Между молекулами амидов, содержащими хотя бы один атом Н при атоме N, возникают водородные связи.
Вследствие частичной
двоесвязанности N=C и, следовательно,
затруднения своб. вращения вокруг связи
С(О)—N амиды могут существовать в цис-
и транс-формах:

В водных р-рах амиды
обычно имеют нейтральную р-цию, что
обусловлено сопряжением своб.
электронной парыатома N
с двойной
связью карбонильной
группы:

Однако первичные и вторичные амиды могут проявлять слабые амфотерные св-ва, а третичные - слабые основные. Так, с сильными минер. к-тами они образуют непрочные легко гидролизующиеся соли. Основные св-ва N-алкилзаме-щенных амидов выражены сильнее, чем у незамещенных. Напр., N,N-диметилацетамид образует с НС1 соль, устойчивую в виде конц. водных р-ров, а с НС1О4 и H2PtCl6 - прочные хорошо кристаллизующиеся соли. В средеуксусного ангидрида амиды количественно оттитровываются р-ром НС1О4. При взаимод. с щелочными металлами у первичных и вторичных амидов атом Н аминогруппы замещается металлом, образуя, напр., RCONHNa.
При кипячении с
конц. водными р-рами минер. к-т
или щелочей амиды
гидролизуются до к-т. Под действием
HNO2легко
дезаминируются: RCONH2 +
HNO2 ->
RCOOH + N2 +
H2O.
Восстанавливаются до аминов RCH2NR'R" натрием в
спиртовой среде, алюмогидридом Li
или Н2 -
над меднохромовыми катализаторами.
Первичные амиды дегидратируются
до нитрилов под
действием Р2О5,
А12О3,
SiO2,
H3PO4 или
др.; при действии гипобромитов илигипохлоритов в
щелочном р-ре превращаются в
первичные амины (Гофмана
перегруппировка). Взаимод. вторичных и
третичных амидов с РС15,
SOC12 и
т.п. приводит к имидоилхлоридам RCC1=NR'
или![]() хлориммониевым солямRCC1—
хлориммониевым солямRCC1—![]() R'R"C1-,
которые при нагревании расщепляют- , ся
на нитрилы RCN
и алкилгалогениды (см. Брауна
реакция).
R'R"C1-,
которые при нагревании расщепляют- , ся
на нитрилы RCN
и алкилгалогениды (см. Брауна
реакция).
С бромом и хлором амиды
образуют соотв. N-бром- и N-хлорамиды,
с формальдегидом и
окисью этилена-N-метилоламиды,
N,N'-метилен-бис-ациламиды и разл.
N-оксиэтильные производные амидов,
имеющие большое пром. значение, напр.:

В пром-сти амиды синтезируют взаимод. к-т или чаще их хлорангидридов, ангидридов, эфиров с NH3 (аммонолиз) либо амином (аминолиз). Еще один пром. способ - неполный гидролиз нитрилов в присуг. H2SO4 или Си: RCN + Н2О -> RCONH2. Разработаны непрерывные контактно-каталитич. аммонолиз и аминолиз к-т при 200-280 С в присут.катализаторов дегидратации (А12О3, SiO2 и др.); выход амидов 95-98%.
В'лаб. условиях
амиды можно синтезировать также
р-цией кетенов с
NH3 или амином (напр.,
СН2=С=О
+ NH3 ->
CH3CONH2),
N-алкилзамещенные амиды-взаимод. амидов
с алкилгалогенидами, N-алкил- и
N-арилзамещенные - с использованием Бекмана
перегруппировки или
перегруппировки Шмидта:

Образование амидов используют для защиты аминогруппы и для идентификации первичных и вторичных аминов(преим. в виде ацетамидов и бензамидов), а также карбоновых к-т (в виде незамещенных амидов, анилидов, бензиламидов). Особое значение методы защиты МН2 - группы имеют в синтезе пептидов (см. Белки).
Амиды - пластификаторы бумаги, искусственной кожи, ПВХ, экстрагенты нек-рых радиоактивных металлов, сырье в произ-ве полимеров, промежут. продукты в синтезе красителей и сульфамидных препаратов и др.
23.2
УГЛЕВОДЫ (сахара), обширная группа полигидроксикарбо-нильных соед., входящих в состав всех живых организмов; к углеводам относят также мн. производные, получаемые при хим. Mодификации этих соед. путем окисления,восстановления или введения разл. заместителей.
Термин "углеводы" возник потому, что первые известные представители углеводов по составу отвечали ф-ле CmH2nOn (угле-род+вода); впоследствии были обнаружены природные углеводы с др. элементным составом.
Классификация и распространение. Углеводы принято делить на моносахариды, олигосахариды и полисахариды.
Моносахариды обычно представляют собой полигид-роксиальдегиды (альдозы) или полигидроксикетоны (кетозы) с линейной цепью из 3-9 атомов С, каждый из к-рых (кроме карбонильного) связан с группой ОН. Простейший моноса-харид, глицериновый альдегид, содержит один асим. атом С и известен в виде двух оптич. антиподов (D и L). Прочиемоносахариды имеют неск. асим. атомов С; их рассматривают как производные D- или L-глицеринового альдегида и относят к D- или L-ряду в соответствии с абс. конфигурацией асим. атома С, наиб. удаленного от карбонильной группы. Различия между изомерными моносахаридами в каждом ряду обусловлены относит, конфигурацией остальных асим. центров.
Характерное св-во моносахаридов в р-рах - мутаротация, т. е. установление таугомерного равновесия между ациклич. альдегидо- или кетоформой, двумя пятичленными (фураноз-ными) и двумя шестичленными (пиранозными) полуацеталь-ными формами. Две пиранозы (как и две фуранозы) отличаются друг от друга конфигурацией (a или b) нового асим. (аномерного) центра, возникающего из карбонильного атома С при циклизации.
Полуацетальный (гликозидный) гидроксил циклич. форм моносахаридов резко отличается от прочих групп ОН моно-сахарида значительно большей склонностью к р-циям нукле-оф. замещения. Такие р-ции приводят к образованию глико-зидов (остаток нуклеофила в гликозиде - напр. спирта. меркаптана - носит назв. агликон). В тех случаях, когдаагликоном служит др. молекула моносахарида, образуются олиго- и полисахариды. При этом каждый остатокмоносахарида может в принципе иметь пиранозную или фуранозную форму, a- или b-конфигурацию гликозидного центра и быть связанным с любой из гидроксильных групп соседнего моносахарида. Поэтому число разл. по строению полимерных молекул, к-рые теоретически можно построить даже из остатков только одногомоносахарида, представляет собой аст-рономич. величину.
К наиб, обычным и распространенным в природе моноса-харидам относят D-глюкозу, D-галактозу, D-маннозу, D-фрук-тозу, D-ксилозу, L-арабинозу и D-рибозу. Из представителей др. классов моносахаридов часто встречаются: 1) дезоксиса-хара, в молекулах к-рых одна или неск. групп ОН заменены атомами H (напр., L-рамноза, L-фукоза, 2-дезокси-D-рибоза); 2) аминосахара, где одна или неск. групп ОН заменены на аминогруппы (напр., 2-амино-2-дезокси-D-глюкоза, или D-глюкозамин); 3) многоатомные спирты (полиолы, альди-ты), образующиеся привосстановлении карбонильной группы моносахаридов (D-сорбит из D-глюкозы, D-маннит из D-маннозы, и др.); 4) уроно-вые кислоты - альдозы, у к-рых группа CH2OH окислена в карбоксильную (напр., D-глюкуроновая к-та); 5) разветвленные сахара, содержащие нелинейную цепь углеродных атомов (напр., апиоза, или 3-С- гидроксиметил-D-глицеро-тетроза; ф-ла I); 6) высшие сахара с длиной цепи более шести атомов С (напр., D-седогеп-тулоза и сиаловые к-ты; ф-лы см. соотв. в статьях Пентозо-фосфатный цикл и Моносахариды}.

За исключением D-глюкозы и D-фруктозы своб. моносахариды встречаются в природе редко. Обычно они входят в состав разнообразных гликозидов, олиго- и полисахаридов и м. б. получены из них после кислотного гидролиза. Разработаны многочисл. методы хим. синтеза редких моносахаридов исходя из более доступных.
Олигосахариды содержат в своем составе от 2 до 10-20 моносахаридных остатков, связанных гликозидными связями. Наиб, распространены дисахариды, выполняющие ф-цию запасных B-B: сахароза в растениях, трегалоза в насекомых и грибах, лактоза в молоке млекопитающих. Известны многочисл. гликозиды олигосахаридов, к к-рым относят разл. физиологически активные в-ва, напр, гликозиды сердечные, нек-рые сапонины (в растениях), мн.антибиотики (в грибах и бактериях), гликолипиды.
Полисахариды- высокомол. соед., линейные или разветвленные молекулы к-рых построены из остатковмоносахаридов, связанных гликозидными связями. В состав полисахаридов могут входить также заместители неуглеводной природы (остатки алифатич. к-т, фосфат, сульфат). В свою очередь цепи высших олигосахаридов иполисахаридов могут присоединяться к полипептидным цепям с образованием гликопротеинов.
Особую группу составляют биополимеры, в молекулах к-рых остатки полиолов, гликозилполиолов, нуклеозидов или моно- и олигосахаридов соединены не гликозидными, а фос-фодиэфирными связями. К этой группе относяттейхоевые кислоты бактерий, компоненты клеточных стенок нек-рых дрожжей, а также нуклеиновые кислоты, в основе к-рых лежит поли-D-рибозофосфатная (РНК) или поли-2-дезок-си-D-рибозофосфатная (ДНК) цепь.
24.1