

Моносахариды
Первые выделенные из природных источников сахара (так же, как и большинство известных в настоящее время) имели химическую формулу Сn (H2 О)n.
Именно поэтому они получили название углеводы. В дальнейшем были получены: - сахара, с другим соотношением углерода и кислорода; - сахара, содержащие другие атомы (азот, серу).
По числу углеродных атомов, входящих в состав молекулы моносахариды делятся на триозы, тетрозы, пентозы, гексозы и т. д. В природе наиболее широко распространены пентозы и гексозы.
Синтез моносахаридов по методу Килиани-Фишера применяется для увеличения длины углеродной цепи альдозы на один атом углерода, в результате чего возникают две диастереомерные альдозы. Уже на первой стадии синтеза образуются изомерные циангидрины, дающие после гидролиза и декгидратации лактоны, которые восстанавливаются до альдоз, содержащих на один атом углерода больше, чем исходный моносахарид.
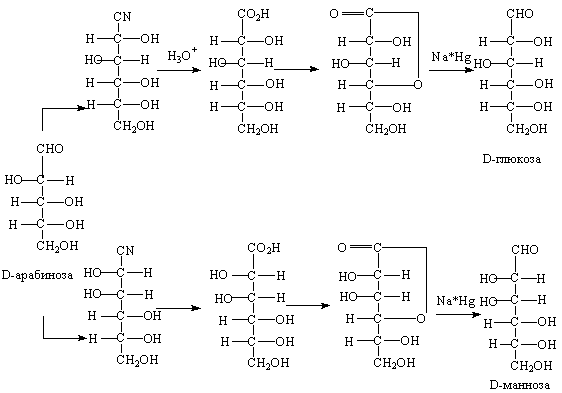
5.2
Соединения характеризуются полярной сопряженной системой. В виду значительного индуктивного (- I) эффекта и большого изомерного (- М) эффекта карбонильной группы двойная углерод – углеродная связь сильно поляризована.

Дипольный момент таких соединений больше, чем для насыщенных карбонильных соединений. ,-Ненасыщенные карбонильные соединения в молекуле имеют два электрофильных реакционных центра.
Химические свойства.
, -ненасыщенные карбонильные соединения могут вступать в обычные реакции присоединения и конденсации по карбонильной группе, такие как образование циангидридов и гидразонов и присоединение металлоорганических соединений. Эти реакции могут осложняться, а иногда перекрываться 1,4–присоединением.
1. Циангидринирование
Реакция циановодорода с альдегидами протекает как 1,2–присоединение, так как этот путь более выгоден с точки зрения скорости и с точки зрения равновесия, чем 1,4–присоединение.

Образование циангидринов кетонов менее выгодно с точки зрения скорости и равновесия и в результате 1,4–присоединение приводит к -цианокетону

Механизм

2. Присоединение воды и галогеноводородов к , - ненасыщенным альдегидам и кетонам

В кислой среде α, β–ненасыщенные альдегиды и кетоны присоединяют воду по связи С=С против правила Марковникова. Однако первичным взаимодействием в этой реакции является 1,4–присоединение.
Аналогично идет присоединение кислот НХ.

3. Реакции нуклеофильного присоединения
а) взаимодействие с Mg–органическими соединениями
Альдегиды и кетоны ведут себя по разному с Mg–органическими соединениями. Альдегиды присоединяют их по >C=O связи, как и насыщенные альдегиды.

Кетоны реагируют по типу 1,4-присоединения.

б) присоединение аммиака и аминов идет по типу 1,4–присоединения, которое в силу неустойчивости интермедиатов превращается в продукт 3,4–присоединения.

в) присоединение триалкилборатов. Эта реакция приводит к образованию -алкил- замещенных насыщенных альдегидов или кетонов.

6.1
реакции с N-нуклеофилами (реакции присоединения-отщепления). К числу N-нуклеофилов (азотистых оснований) относятся аммиак NН3 и ряд его производных с общей формулой NH2–X, например, NH2–R, NH2–NH2, NH2–NH–Аr, NH2–ОН и др. Обычно используют кислые катализаторы для активации субстрата и дегидратации аддукта. Однако в сильнокислой среде азотистые основания превращаются в соли и теряют нуклеофильные свойства, поэтому каждую конкретную реакцию следует проводить при оптимальном значении рН среды. Учитывая кислый катализ, механизм взаимодействия карбонильного соединения с азотистым основанием можно представить следующим образом:

Как видно из схемы, на лимитирующей стадии протекает типичная для карбонильных соединений AN реакция. Однако промежуточный аддукт неустойчив, так как энергия двух простых связей C–N и С–О (305,9 + 356,2 = 662,1 кДж/моль) больше энергии двойной связиC=N (615,9 кДж/моль). Следовательно, энергия аддукта относительно велика и поэтому легко протекает его дегидратация. Таким образом, реакции карбонильных соединений с N-нуклеофилами можно рассматривать как нуклеофильное замещение атома кислорода карбонильной группы (SN).
2.1 Образование иминов и оксимов:

При реакции самого аммиака с карбонильными соединениями образуются неустойчивые и склонные к полимеризации имины. Имины, содержащие хотя бы один ароматический заместитель у карбонильного углерода (Аr–СН=N–R), являются устойчивыми продуктами. Они известны как основания Шиффа (часть 2, глава 2).
2.1.1Гидролиз иминов. Имины легко гидролизуются в кислой или щелочной среде, что позволяет отделить карбонильные соединения от соединений других классов и выделить их в чистом виде:

2.1.2Гидролиз оксимов. Оксимы, как и многие производные карбонильных соединений, в водных растворах кислот могут подвергаться гидролизу, регенерируя исходное карбонильное соединение и гидроксиламин. В присутствии сильных кислот альдоксимы могут подвергаться дегидратации:

7.1
a-Дикарбонильные
соединения (1,2-дикарбонильные соединения)
RCOCOR'. Карбонильные
группы обычно обнаруживают ярко
выраженную электроф. реакц. способность.
Для них характерна циклизация при
действии бифункциональных нуклеофилов
(напр., ур-ние 1) и соед. 3-валентного Р
(2). Связь С—С между группами СО обычно
сравнительно легко расщепляется, напр.,
при фотолизе (3)
или действии нуклеофилов (4). Для a-диальдегида
(глиоксаля)и a-кетоальдегидов
характерна внутримол. Канниццаро
реакция,
для ароматич. a-дикетонов
(бензилов)
- бензиловая
перегруппировка.
 Осн.
способы получения a-дикарбонильных
соединений - окисление монокарбонильных
соед. (5) или их a-замещенных
производных, напр.ацилоинов (6),
производных ендиолов (7), гликолей и
др.; ароматич. a-дикетоны
получают также из ароматич. соед. и
оксалилхлорида по р-ции Фриделя -
Крафтса.
Осн.
способы получения a-дикарбонильных
соединений - окисление монокарбонильных
соед. (5) или их a-замещенных
производных, напр.ацилоинов (6),
производных ендиолов (7), гликолей и
др.; ароматич. a-дикетоны
получают также из ароматич. соед. и
оксалилхлорида по р-ции Фриделя -
Крафтса.
 Нек-рые a-дикарбонильные
соединения, напр., метилглиоксаль
СН3СОСНО,
пировиноградная к-та СН3СОСООН,
участвуют в обмене в-в в
живыхорганизмах. Диацетил обусловливает
запах сливочного масла;
его производное - диметилглиоксим применяется
как аналит. реагент. a-Дикарбонильные
соединения используют в орг. синтезе
для получения гетероциклич. соед.
Нек-рые a-дикарбонильные
соединения, напр., метилглиоксаль
СН3СОСНО,
пировиноградная к-та СН3СОСООН,
участвуют в обмене в-в в
живыхорганизмах. Диацетил обусловливает
запах сливочного масла;
его производное - диметилглиоксим применяется
как аналит. реагент. a-Дикарбонильные
соединения используют в орг. синтезе
для получения гетероциклич. соед.
7.2
Классификация гетероциклических соединений.
Гетероциклические соединения – это органические вещества, содержащие в своих молекулах циклы, в образовании которых кроме атомов углерода участвуют атомы других элементов (гетероатомы).
Размер цикла может быть разным. Наиболее распространены в природе пяти и шестичленные циклы, в состав которых входят атомы азота, кислорода или серы.
В зависимости от природы гетероатома различают нитроген-, оксиген- и серосодержащие циклы. Их также различают по числу гетероатомов, числу звеньев в цикле и других признаках.
По степени насыщенности все гетероциклические соединения могут быть насыщенными, ненасыщенными и ароматическими.
|
Простые гетероциклы с одним гетероатомом | ||||||||
|
|
Насыщенные гетероциклы |
Ненасыщенные гетероциклы | ||||||
|
Гетероатом |
Азот |
Кислород |
Сера |
Азот |
Кислород |
Сера |
| |
|
Трёхчленные |
| |||||||
|
Систематическое название |
Азиридин |
Оксиран |
Тииран |
Азирин |
Оксирен |
Тиирен |
| |
|
Тривиальное название |
Этиленимин |
Этиленоксид |
Этиленсульфид |
- |
- |
- |
| |
|
Структура |
|
|
|
|
|
|
| |
|
Четырёхчленные |
| |||||||
|
Систематическое название |
Азетидин |
Оксетан |
Тиетан |
Азет |
Оксет |
Тиет |
| |
|
Тривиальное название |
1,3-Пропиленимин |
Триметиленоксид |
Триметиленсульфид |
Азациклобутадиен |
- |
- |
| |
|
Структура |
|
|
|
|
|
|
| |
|
Пятичленные |
| |||||||
|
Систематическое название |
Азолидин |
Оксолан |
Тиолан |
Азол |
Оксол |
Тиол |
| |
|
Тривиальное название |
Пирролидин |
Тетрагидрофуран |
Тетрагидротиофен |
Пиррол |
Фуран |
Тиофен |
| |
|
Структура |
|
|
|
|
|
|
| |
|
Шестичленные |
| |||||||
|
Систематическое название |
Азинан |
Оксан |
Тиан |
Азин |
Оксиний |
Тииний |
| |
|
Тривиальное название |
Пиперидин |
Тетрагидропиран |
Тетрагидротиопиран |
Пиридин |
Пирилий |
Тиопирилий |
| |
|
Структура |
|
|
|
|
|
|
| |
|
Семичленные |
| |||||||
|
Систематическое название |
Азепан |
Оксепан |
Тиепан |
Азепин |
Оксепин |
Тиепин |
| |
|
Тривиальное название |
Гексаметиленимин |
Гексаметиленоксид |
Гексаметиленсульфид |
Азатропилиден |
Оксациклогептатриен |
- |
| |
|
Структура |
|
|
|
|
|
|
| |




























|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
8.1
ВИТТИГА РЕАКЦИЯ, получение олефиновдействием илидов Р (алкилиденфосфоранов) наальдегидыиликетоны.Илидыобычно используют в свежеприготовленном виде. Получают их взаимод. трифенилалкилфосфония с литийорг. соед. или гексаалкилтриамидоалкилфосфония сощелочами.
Механизм
Виттига реакцииможно
представить след. образом:
Обычно образуется смесь изомерных олефинов. Однако при соответствующем подборереагентови условий р-ции (напр., при использовании полярных апротонных р-рителей) возможен синтез преим. одного изизомеров(цис- или транс-изомера).
Виттига реакциюшироко
используют в тонком орг. синтезе, напр.
для полученияальдегидов,
содержащих на одинатомС
больше, чем в исходном соед., а
такжеполиенов,
гетероциклич. соед.: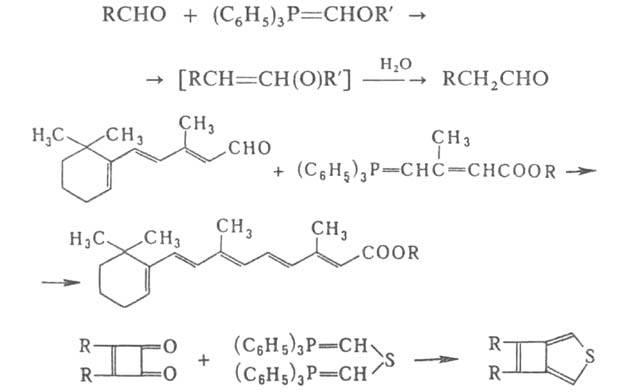
Р-ция открыта Г. Виттигом в 1954.
9.1
Взаимодействие со спиртами. Альдегиды могут взаимодействовать с одной или двумя молекулами спирта, образуя соответственно полуацетали и ацетали.
Полуацеталями называют соединения, содержащие при одном атоме углерода гидроксильную и алкоксильную (OR) группы. Ацетали — это соединения, содержащие при одном атоме углерода две алкоксильные группы:

полуацеталь ацеталь
Реакцию получения ацеталей широко используют в органических синтезах для "защиты" активной альдегидной группы от нежелательных реакций:
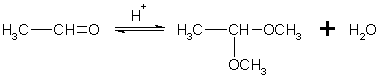
Особенно важное значение подобные реакции имеют в химии углеводов.
Низшие ацетали — жидкости приятного фруктового запаха, растворимые в органических растворителях и нерастворимые в воде.
Ацетали устойчивы в щелочных средах, однако в присутствии кислот легко гидролизуются водой с образованием альдегидов и спиртов, производными которых они являются. Полуацетали, как правило, ещё легче подвержены гидролизу и в растворах находятся в равновесии с исходным альдегидом и спиртом.
Такое поведение ацеталей и гемиацеталей — как и их образование из карбонильных соединений — обусловлено общим механизмом кислотного катализа с образованием резонансно стабилизированного α-атомом кислорода карбокатиона:
R-CHO
+ H+ ![]() R-СH+-OH
R-СH+-OH ![]() R-СH=O+H
R-СH=O+H
R-СH+-OH
+ ROH ![]() R-CH(OR)OH
+ H+
R-CH(OR)OH
+ H+
R-CH(OR)OH
+ H+ +
H+ ![]() R-СH+-OR
+ H2O
R-СH+-OR
+ H2O
R-СH+-OR
+ ROH ![]() R-CH(OR)2 +
H+
R-CH(OR)2 +
H+
Все эти реакции обратимы, и положение равновесия может быть сдвинуто удалением из реакционной смеси какого-либо компонента. Другим следствием обратимости реакций ацеталей в условиях кислотного катализа, идущих с образованием карбокатионов, являются реакции переацетализации:
R-CH(OR)2 +
H+ ![]() R-СH+-OR
R-СH+-OR ![]() R-СH=O+R
R-СH=O+R
R-СH+-OR
+ R1XH ![]() R-CH(OR)XR1
R-CH(OR)XR1
X = O, S,
Переацетализация происходит при взаимодействии ацеталей не только со спиртами, но и при реакции с тиолами и ведет в последнем случае к образованиюдитиоацеталей R2C(SR)2.
В реакциях с азотистыми нуклеофилами ацетали выступают как функциональные аналоги исходных карбонильных соединений: так, в реакциях ацеталей с первичными аминами, гидразинами и гидроксиламином образуются имины, гидразоны и оксимы:
R2C(OR1)2 +
R2NH2 ![]() R2C=NR2 +
2 R1OH
R2C=NR2 +
2 R1OH
R2 = Alk, Ar (имины), NR2 (гидразоны), OH (оксимы)
При взаимодействии вторичных аминов с ацеталями — функциональными аналогами енолизирующихся альдегидов и кетонов образуются енамины:
R12CHCR2(OR)2 +
R3NH ![]() R12C=CR2NR3
R12C=CR2NR3
10.1
МАННИХА РЕАКЦИЯ, аминометилирование соед.
с подвижным атомом водорода под
действием формальдегида и
в осн. вторичных аминов с
образованием т.
наз. оснований Манниха:
![]() Первичные амины и аммиак реагируют
аналогично, однако образующиеся продукты
способны к дальнейшим превращениям. Обычно
Манниха реакцию проводят
в присут. к-т, реже - оснований.
Наиб. используемые р-рители -спирты,
Н2О,
СН3СООН, нитробензол.
В качестве соед., содержащих подвижный атом Н,
используют, как правило,альдегиды, кетоны,
карбоновые к-ты и их производные, фенолы,
нитроалканы, гетероциклич. соед.,
производныеацетилена,
синильную к-ту, напр.:
Первичные амины и аммиак реагируют
аналогично, однако образующиеся продукты
способны к дальнейшим превращениям. Обычно
Манниха реакцию проводят
в присут. к-т, реже - оснований.
Наиб. используемые р-рители -спирты,
Н2О,
СН3СООН, нитробензол.
В качестве соед., содержащих подвижный атом Н,
используют, как правило,альдегиды, кетоны,
карбоновые к-ты и их производные, фенолы,
нитроалканы, гетероциклич. соед.,
производныеацетилена,
синильную к-ту, напр.:
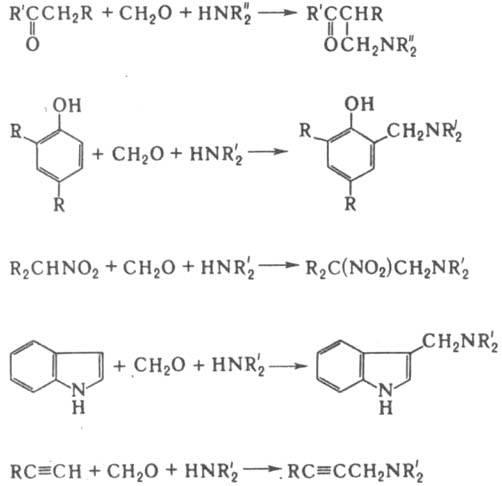 В
нек-рых случаях вместо СН2О
применяют алифатич. и ароматич. альдегиды и кетоны.
Диальдегиды вступают в
двойную конденсацию Манниха,
напр., в синтезе алкалоида тропинона:
В
нек-рых случаях вместо СН2О
применяют алифатич. и ароматич. альдегиды и кетоны.
Диальдегиды вступают в
двойную конденсацию Манниха,
напр., в синтезе алкалоида тропинона:
 При
использовании в качестве соед. с
подвижным атомом Н спиртов, аминов, тиолов, фосфинов и
др. р-ция протекает соотв. как О-, N-, S- или
Р-аминометилирование, напр.:
При
использовании в качестве соед. с
подвижным атомом Н спиртов, аминов, тиолов, фосфинов и
др. р-ция протекает соотв. как О-, N-, S- или
Р-аминометилирование, напр.:
RОН + СН2О + HNR'2 : ROCH2NR'2;
R2NH + СН2O + HNR'2 : R2NCH2NR'2;
PH3 + CH2O + HNR2 : P(CH2NR2)3
Предполагают,
что Манниха реакция протекает
через иминиевые
соли,
образующиеся из СН2О
и амина и
реагирующие затем с образованием
аминопроизводного, напр.:
 Иногда
в Маннихареакцию вводят
заранее приготовленную соль иминия,
напр. CH2=NCH3 Cl-,
что облегчает
проведение аминометилирования. Основания Манниха
- промежут. продукты при получении
ненасыщ. кетонов,альдегидов, нитросоединений и
гетероциклов. Р-ция применяется в
синтезе прир. в-в и лек. препаратов.
Открыта К. Маннихом в 1917.
Иногда
в Маннихареакцию вводят
заранее приготовленную соль иминия,
напр. CH2=NCH3 Cl-,
что облегчает
проведение аминометилирования. Основания Манниха
- промежут. продукты при получении
ненасыщ. кетонов,альдегидов, нитросоединений и
гетероциклов. Р-ция применяется в
синтезе прир. в-в и лек. препаратов.
Открыта К. Маннихом в 1917.
10.2
АЦЕТОУКСУСНЫЙ
ЭФИР (этиловый
эфир ацетоуксусной
к-ты, этилацетоацетат), мол. м. 130,1418;
бесцв.жидкость;
т. пл. -45°С, т. кип. 180,8°С (с разл.), 100°С/80
мм рт. ст., 71°С/12,5мм рт.ст.; d420 1,0284,
nD20 1,4198;
раств. в воде (14,3%
при 16,5°С), этаноле,
эфире. Существует в двух таутомерных
формах - кетонной (а)и енольной (б):
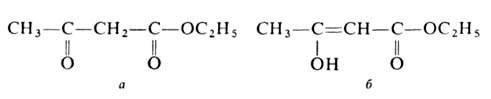
Енольная форма стабилизируется за счет сопряжения двойной углерод-углеродной связи с карбонильной группой и образования внутримолекулярной водородной связи между гидроксилом и карбонилом. Кетонную форму можно выделить вымораживанием, енольную - фракционной перегонкой в кварцевой посуде в вакууме; для них т. пл. соотв. -39 и -44°С, d420 1,0368 и 1,0119, nD20 1,4425 и 1,4480. Соотношение таутомеров зависит от природы р-рителя, материала сосуда, т-ры, напр. в ацетоуксусном эфире при комнатной т-ре содержится 7,5% енола, при 18°С в р-рах ацетоуксусног эфира в воде, этаноле, эфире и циклогексане - соотв. 0,4, 12, 27,1 и 46,4%. Чистые таутомерные формы сохраняются в кварцевой посуде при -80°С.
Под
действием разб. р-ров к-т
или щелочей ацетоуксусный
эфир подвергается расщеплению с
образованиемацетона (т.
н. кетонное расщепление), под действием
конц. р-ров щелочей -
с образованием уксусной к-ты (кислотное
расщепление):

Как кетон ацетоуксусный
эфир восстанавливается Н2 in
situ до этил![]() гидроксибутирата;
присоединяет, напр., HCN, NaHSO3 с
образованием соотв. циангидрина и
гидросульфитного производного. Р-ция
с фенилгидразиномсопровождается циклизацией фенилгидразона
ацетоуксусного эфира:
гидроксибутирата;
присоединяет, напр., HCN, NaHSO3 с
образованием соотв. циангидрина и
гидросульфитного производного. Р-ция
с фенилгидразиномсопровождается циклизацией фенилгидразона
ацетоуксусного эфира:

Как енол ацетоуксусный
эфир мгновенно обесцвечивает р-р
Вг2 (р-ция
служит для количеств. определения енола),
с Fed, образует комплекс красно-фиолетового
цвета, с РС15-![]() хлоркротоновый
эфир СН3СС1=СНСООС2Н5.
Ацетилируется хлорангидридами к-т:
хлоркротоновый
эфир СН3СС1=СНСООС2Н5.
Ацетилируется хлорангидридами к-т:

При действии на ацетоуксусный эфир металлич. Na или алкоголятов Na в спиртовом р-ре получается натрийацетоуксусный эфир CH3C(ONa)—СНСООС2Н5, широко используемый в р-циях с алкилгалогенидами, приводящих к образованию алкил- и диалкилацетоуксусных эфиров-соответственно CH3COCH(R)COOC2H5 и CH3COC(RR')COOC2H5. Кетонное и кислотное расщепление последних происходит так же, как и ацетоуксусного эфира. Напр., при кетонном расщеплении метилацетоуксусного эфира получается метилэтилкетон, при кислотном - уксусная и пропионовая к-ты. В зависимости от характера реагента, природы р-рителя и условий из натрийацетоуксусного эфира могут получаться как С-, так и О-производные ацетоуксусного эфира.
Атомы Н
метиленовой группы ацетоуксусного
эфира очень подвижны, благодаря чему
он реагирует, напр., сальдегидами:

вступает в р-цию Михаэля и др. В пром-сти и лаборатории ацетоуксусный эфир получают действием металлич. Na на этилацетат (см. Клайзена конденсация]. Образующийся натрийацетоуксусный эфир действием разб. минер. к-т переводят в ацетоуксусный эфир. Применяют ацетоуксусный эфир в синтезе лек. ср-в (напр., амидопирина,антипирина, акрихина), витамина В1, красителей, содержащих пиразолоновый цикл; как ароматизирующее в-во для пищ. продуктов. Ацетоуксусный эфир раздражает кожу. Т. всп. 85оС.
11.1
МEЕРВЕЙНА - ПОННДОРФА - ВЕРЛEЯ РЕАКЦИЯ, восстановлениеальдегидови кетонов доспиртовв присут.алкоголятовАl:
![]()
Р-цию чаще всего проводят нагреванием карбонильного соед. с изопропилатом Аl в р-ре безводного изопропанола, реже-бензола илитолуола. Р-ция обратима (обратное превращ. наз.Оппенауэра реакцией),равновесиесдвигается вправо при непрерывной отгонке образующегосякетона. Для увеличения скорости р-ции и уменьшения количествапобочных продуктов обычно используют избыток алкого-лята Аl.
В р-цию вступают алифатич. и ароматич. карбонильные соединения.Альдегидывосстанавливаются легче кетонов. Электронодонорные заместители увеличивают скорость р-ции. Привосстановлениикетонов, содержащихаминогруппы, к-рые могут образовывать комплекс ссолямиАl, более эффективным оказываетсявосстановлениев присут. пропилата Na. Длявосстановленияa-бромкетонов в бром-гидрины (протекающего при низких т-рах) рекомендуется использовать AlCl2OR. Меервейна - Понндорфа - Верлея реакцияселективна, связи С=С, сложноэфирные и нитрогруппы, а также галогензаместителивосстановлениюне подвергаются.
Предполагают, что Меервейна - Понндорфа - Верлея реакцияосуществляется через образование комплекса между изопропилатом Аl и карбонильным соед., гидридное перемещение, элиминированиеацетонаиз комплекса иалкоголизсмешанногоалкоголята:
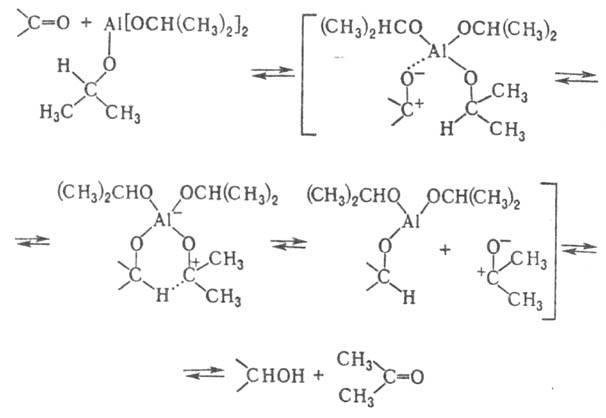
Гидрид-ионприсоединяется к карбонильному соед. обычно с пространственно менее затрудненной стороны. Так, привосстановлениициклогексаноновосновной продукт р-ции -соответствующий аксиальныйспирт. Наряду свосстановлениемпо вышеуказанному механизму, может протекать конкурирующая р-ция с образованием переходного состояния, включающего двамоляалкоголята.
Модификация Меервейна - Понндорфа - Верлея реакции-высокоселективное восстановлениеальдегидовспиртомв присут. безводного Аl2О3, напр.:

ТИЩЕНКО РЕАКЦИЯ
(Тищенко-Клайзена р-ция), диспропорционирование альдегидов под действием алкого-лятов А1 собразованием сложных эфиров:
![]()
В р-цию вступают ароматич., алифатич. и нек-рые гетеро-циклич. альдегиды. Из алкоголятов Аl в Т. р.обычно используют этилат, изопропилат или бутилат Аl; иногда их активируют добавками АlСl3, ZnO, HgCl2,Zn(OR:)2 или Mg(OR:)2. Р-ция также катализируется комплексами Ru или V, борной к-той,тетракарбонилферратом Na-Na2[Fe(CO)4] (последний используют для конденсации ароматич. альдегидов).В случае ароматич. альдегидов можно использовать также алкоголяты Na и К.
Р-цию обычно проводят в отсутствие р-рителя (реже-в инертном орг. р-рителе) при комнатной т-ре. Смесьальдегида и алкоголята А1 выдерживают неск. часов. Выходы 50-95%.
При использовании двух разл. альдегидов наблюдается т. наз. перекрестная Т. р., напр.:

Альдегиды и катализатор можно подобрать таким образом, что в преобладающем кол-ве будетобразовываться один из двух продуктов перекрестной Т. р.
Осн. побочные продукты-гидроксиальдегиды и ненасыщ. карбонильные соед., образующиеся в результатеальдольной и кротоновой конденсаций. В преобладающем кол-ве эти соед. образуются в присут.алкоголятов Na и К из алифатич. альдегидов, имеющих атом H в a-положении к карбонильной группе. Прииспользовании в качестве катализаторов алкоголятов Са или Mg процесс альдольной конденсации и Т. р.часто совмещаются, напр.:

В присут. Н 2 О обычно увеличивается выход побочных продуктов (в т. ч. в результате гидролиза алкоголятаАl). Однако в ряде случаев (напр.; при проведении р-ции с нек-рыми пространственно-затрудненнымиальдегидами) Т. р. проводят в присут. Н 2 О, но без катализатора, напр.:

(выход 90%)
Детально механизм Т. р. не исследован. Установлено, что ключевая стадия р-ции-межмол. перенос гидрид-иона, подобно тому как это происходит в Канниццаро реакции. Предполагают, что перенос гидрид-ионаосуществляется внутри комплекса двух молекул альдегида с алкоголятом Аl.
11.2
Ацетаты – это соли и эфиры уксусной кислоты. Соли – кристаллические продукты большинство хорошо растворимы в воде. Многим находят разнообразное применение. Эфиры – это летучие жидкости с фруктовыми и цветочными запахами.
Ацетоуксусный эфир, бесцветная жидкость с приятным запахом, температурой кипения 181°, с частичным разложением; плотностью 1028,2 кг/м³.
Ацетоуксусный эфир существует в двух таутометрических формах: кетонной СН3С(О)СН2СООС2Н5 и емольной СН3С(ОН)=СНСООС2Н5.
Ацетоуксусный эфир расщепляется при действии на него щелочей. Это расщепление в зависимости от условий происходит различно. При действии разбавленных растворов щелочей (или кислот) ацетоуксусный эфир омыляется с последующим отщеплением СО2 от образовавшейся ацетоцуксусной кислоты:
СН3СОСН3-СО-С-С2Н5+Н-О-Н→СН3СОСН2-СО-ОН+С2Н5О4
СН3СОСН2-СО-О-Н→СН3СОСН3+СО2
Этот вид расщепления ацетоуксусного эфира называется кетонным расщеплением. В результате его образуется двуокись углерода, этиловый спирт и ацетон.
При нагревании ацетоуксусного эфира с концентрированными растворами щелочей происходит кислотное расщепление, и образуются две молекулы уксусной кислоты:
При действии на натрийацетоуксусный эфир алкилгалогениодов алкил присоединяется к атому углерода, не смотря на то, что, в исходном натрийацетоуксусном эфире натрий был связан с атомом кислорода:
Н3С – С = СН – СООС2Н5 +1-R →
СН3-СО-СН-СООС2Н5+Na!
В полученном моноалкимерованном эфире оставшийся атом водорода может быть заменен натрием, который, в свою очередь, можно тем же путем заместить еще одним радикалом:
В результате получаются двузамещенные производные ацетоуксусного эфира. Подобно самому ацетоуксусному эфиру, его одно- и двузамещенные производные способны подвергаться кетонному расщеплению. Это позволяет синтезировать кетоны, у которых один из радикалов – метил, а другой может иметь нормальную или разветвленную цепь:
СН3ССНR – СООС2Н5 →СН3 – С – СН2 – R
СН3ССНRR’ – СООС2Н5 →СН3 – С – СН
Из ацетоуксусного эфира и его одно- и двузамещенных производных можно, используя кислотное расщепление, синтезироватьт также и кислоты нормального строения:
СН3ССНR – СООС2Н5 → RСН2 СООН
СН3ССНR – СООС2Н5 → СН СООН
До того, как были разработаны методы синтеза на основе малоновой кислоты, расщепление замещенных ацетатуксусных эфиров имело важное значение для синтеза карбоновых кислот; в некоторых случаях этот метод используется и сейчас. Реакция, обратная конденсации Клейзена, до некоторой степени происходит даже при гидролизе разбавленной щелочью, и поэтому карбоновые кислоты образуются как побочные продукты:
СН3СОСН2СООС2Н5 →СН3СОО- + СН3СОО- - С2Н5ОН
12.1
Реакции окисления.
По отношению к различным окислителям свойства альдегидов и кетонов сильно различаются. Большинство окислителей, включая воздух, легко окисляют альдегиды до кислот. Особенно легко окисляются ароматические альдегиды.
 (75)
(75)
Реакция проходит по радикальному механизму через образование гидроперекисей. Для сохранения альдегидов от окисления атмосферным воздухом к ним прибавляют небольшие количества антиоксидантов, блокирующих свободные радикалы. В качестве антиоксидантов используют ароматические амины и фенолы.
![]() (76)
(76)
гексаналь гексановая кислота
А. Реакция серебряного зеркала
Легкая окисляемость альдегидов используется для их качественного определения. Окисление альдегидов с помощью растворов, содержащих двухвалентную медь (реактив Фелинга) или серебро (реактив Толленса) является тестом на присутствие альдегида.
![]()
В технике эта реакция используется для серебрения зеркал и игрушек:
![]() (77)
(77)
 (78)
(78)
ванилин ванилиновая кислота
Б. Окисление кетонов
Кетоны значительно более устойчивы к окислению, чем альдегиды, т. к. рядом с карбонильной группой у них нет атома водорода. Кетоны не восстанавливают ни реактив Фелинга ни реактив Толленса. Сильные окислители, такие как перманганат калия и азотная кислота окисляют кетоны
|
| |
|
|
|

с разрывом углеродной цепи до кислот:
Окислением циклогексанона азотной кислотой в промышленности получают адипиновую кислоту:
 (79)
(79)
циклогексанон адипиновая кислота
В. Окисление по Баеру-Виллегеру
Альдегиды и кетоны окисляются надкислотами (окисление по Баеру-Виллегеру), например:
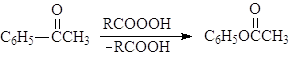 (89)
(89)
ацетофенон фенилацетат
Из двух заместителей карбонильной группы к кислороду прейдет наиболее склонный к миграции. Склонность к миграции уменьшается в следующем ряду:
Н > Ph > 3o алкил > 2o алкил > 1o алкил > метил.
Механизм реакции Баера-Виллегера:
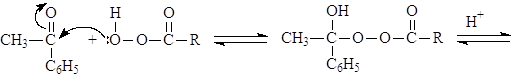 (М
10)
(М
10)

12.2
Дикарбоновые кислоты
Дикарбоновыми (или по-другому двухосновными) называют карбоновые кислоты, которые содержат две карбоксильные группы:За счет наличия дополнительной полярной карбоксильной группы (COOH), способной образовывать водородные связи, дикарбоновые кислоты лучше растворяются в воде и имеют более высокие температуры плавления, чем соответствующие монокарбоновые кислоты.Карбоксильная группа проявляет сильные электроноакцепторные свойства, и поэтому дикарбоновые кислоты являются более сильными кислотами, чем соответствующие монокарбоновые.
Названия дикарбоновых кислот по систематической номенклатуре образуют прибавлением окончания «диовая кислота» к названию алкана. Часто применяют тривиальные названия.
HOOC-COOH этандиовая кислота (щавелевая кислота)
HOOC-CH2 -COOH пропандиовая кислота (малоновая кислота)
Свойства дикарбоновых кислот похожи на свойства монокарбоновых кислот, за некоторыми исключениями, которые особенно проявляются для первых членов гомологического ряда. Начиная с адипиновой кислоты, никаких особенных аномалий уже не наблюдается.
Свойства дикарбоновых кислот
Двухосновные карбоновые кислоты проявляют свойства, характерные для одноосновных кислотам - они образуют соли, сложные эфиры, хлорангидриды и амиды. Помимо этого, они способны вступать в специфические реакции, например, образование циклических ангидридов, двух различных рядов сложных эфиров (кислых и полных) и смешанных производных.
Дикарбоновые кислоты имеют две константы диссоциации, соответствующие каждой из двух карбоксильных групп. С увеличением количества углеродных атомов между группами и становятся практически равными и стремятся к соответствующей одноосновной кислоты. Однако, когда карбоксигруппы расположены рядом, их константы существенно отличаются друг от друга. Это обусловлено тем, что когда отрывается первый протон, молекула становится заряженным ионом, и ей уже гораздо сложнее отдать второй атом водорода.
По физическим свойствам двухосновные кислоты подобны одноосновным. Это кристаллические вещества с высокими температурами плавления. Одной из особенностей дикарбоновых кислот является то, что кислоты с четным числом углеродных атомов плавятся при более высоких температурах, нежели с нечетным (стоит заметить, что с удлинением углеродной цепи разница в температурах постепенно уменьшается). Низшие гомологи хорошо растворяются в воде. Так же, как и монокарбоновые кислоты, двухосновные кислоты способны образовывать межмолекулярные водородные связи.
13.1
Реакция Гриньяра — металлорганическая химическая реакция, в которой арил- или алкилмагнийгалогениды (также называемые реактивами Гриньяра) действуют какнуклеофилы, атакуя электрофильный атом углерода с образованием углерод — углеродной связи. Реакция Гриньяра — важный метод создания углерод-углеродных связей, а также связей углерод-гетероатом (P, Sn, B, Si и др.)