

Галогенангидриды карбоновых кислот
АБВГДЕЖЗИКЛМНОПРСТУФХЦЧШЩЭЮЯ
ГАЛОГЕНАНГИДРИДЫ КАРБОНОВЫХКИСЛОТ (ацилгалогениды), орг. соед. общей ф-лы RC(O)Hal. Жидкие или твердые в-ва (см. табл.), устойчивые в нормальных УС-уловиях (у муравьиной к-ты устойчив только фторангидрид).Галогенангидриды карбоновых кислот -сильные электроф. агенты; их электрофильность возрастает в ряду COF < COC1 < СОВr < COI, однако практич. значение имеют в осн. хлорангидриды и в меньшей степени-бромангидриды. При взаимод, галогенангидридов карбоновых кислот с нуклеофилами образуются продукты ацилирования последних:
RCOHal + НА -> RCOA + HHal
где А = ОН, OR, NH2, NHR, NR2, SH, SR, CN.
ХАРАКТЕРИСТИКИ НЕКОТОРЫХ ГАЛОГЕНАНГИДРИДОВ КАРБОНОВЫХ КИСЛОТ
В присут. к-т
Льюиса галогенангидриды карбоновых
кислот ,
дающие с ними комплексы, ацилируют
ароматич. и гетероароматич. соединения
с образованием кетонов (р-ция Фриделя
- Крафтса), присоединяются
к олефинам иацетиленам,
образуя соотв.![]() гало-генкетоны
и
гало-генкетоны
и![]() галогенвинилкетоны
(р-ция Кондакова). Ввиду
нестойкостигалогенангидридов муравьиной
к-ты ароматич. углеводороды формилируют
смесью СО с НС1, образующими хлористый
формил in situ (р-ция Гаттермана - Коха).
галогенвинилкетоны
(р-ция Кондакова). Ввиду
нестойкостигалогенангидридов муравьиной
к-ты ароматич. углеводороды формилируют
смесью СО с НС1, образующими хлористый
формил in situ (р-ция Гаттермана - Коха).
Галогенангидриды карбоновых
кислот восстанавливаются
Н2 на
палладиевом или
никелевом катализаторе вальдегиды (р-ция
Розенмунда), комплексными гидридами металлов-в спирты.
При действии
третичных аминов нагалогенангидриды карбоновых
кислот ,
имеющие в![]() положении
хотя бы один атом Н,
образуются кетены,
что используется, в частности, при
получении макроциклич. кетонов из
дигалогенангидридов дикарбоновых к-т.
О р-ции с диазометаном см.
Арндта- Аисте рта реакция.
положении
хотя бы один атом Н,
образуются кетены,
что используется, в частности, при
получении макроциклич. кетонов из
дигалогенангидридов дикарбоновых к-т.
О р-ции с диазометаном см.
Арндта- Аисте рта реакция.
Получают галогенангидриды карбоновых кислот действием на карбоновые к-ты или их ангидриды тионилхлорида или тионилбромида (иногда в присут. ДМФА или гексаметилфосфотриамида), галогенидов Р, хлорсульфоновой к-ты. Если к-та (напр., непредельная) чувствительна к действию выделяющегося в этих р-циях HHal, используют р-цию стрифенилфосфином в р-ре СС14 (для получения хлорангидридов) или с др. галогенангидридами, напр. оксалилхлоридом. Применяется также введение группы COHal в непредельные соед. с помощью фосгена,карбонилирование галогензамещенных углеводородов, частичный гидролиз соед., содержащих трихлорметильную группу, и хлорирование ароматич. альдегидов хлоридом серы S2C12. Бром-, иод- и фторангидриды получают также действием на хлорангидриды соответствующих галогеноводородов или их солей.
Для обнаружения и количеств. определения галогенангидриды карбоновых кислот превращают в карбоновые к-ты или их производные (амиды, эфиры, гидроксамовые к-ты), к-рые и анализируют.
Р-цией дигалогенангидридов дикарбоновых к-т с диаминами и дигидроксисоединениями (напр., гликолями, двухатомными фенолами) получают соотв. полиамиды и сложные полиэфиры.
Галогенангидриды карбоновых кислот сильно раздражают слизистые оболочки и кожу. Для галогенангидридовнизших алифатич. карбоновых к-т ПДК 0,1-0,3 мг/м3, для бензоилхлорида-5,0 мг/м3.
Соединения, которые можно рассматривать как продукты, образующиеся при отщеплении одной молекулы воды от двух молекул одноосновной карбоновой к-ты, причем такие ангидриды м. б. симметричными, или несимметричными.
Названия ангидридов (А.) образуют от названий к-т; в случае несимметричных А. в алфавитном порядке перечисляют назв. обеих к-т, напр. (СН3СО)2О - уксусный ангидрид, СН3СО-О-СОС2Н5 - пропионовоуксусный ангидрид.
Ангидриды низших кислот - жидкости. Циклич. ангидриды - кристаллы. Ангидриды - сильные электроф. агенты; с нуклеофилами образуют продукты ацилирования, в случае циклич. ангидридов - продукты, содержащие омега-карбоксильную группу.
Наиболее сильные ацилирующие св-ва - у несимметричных (смешанных) А. незамещенных карбоновых к-т с трифторуксусной CF3COOH или трифторметансульфоновой CF3SO3H к-тами. Эти ангидриды, часто получаемые in situ из карбоновых к-т и ангидридов фторсодержащих к-т, ацилируют бензол в отсутствие катализаторов. При конденсации ароматических альдегидов с ангидридами алифатич. карбоновых к-т образуются альфа-замещенные коричные к-ты (р-ция Перкина): АгСНО + (RCH2CO)2O --> ArCH=C(R)COOH. Циклич. А. непредельных к-т (малеиновой, тетрагидрофталевой) реагируют как диенофилы в диеновом синтезе.
Восстановлением циклич. ангидридов комплексными гидридами металлов получают лактоны.
Способы получения
Ангидриды могут быть получены действием на кислоты водоотнимающими средствами - Р2О5, тионилхлоридом SOC12, фосгеном. Муравьиная к-та образует только смешанные А., являющиеся формилирующими агентами. При нагр. двухосновных высших жирных к-т (не менее С6) с уксусным ангидридом образуются полиангидриды, напр, адипиновый НО[-СО(СН2)4СОО-]nН.
24.2
Белки́ (протеи́ны, полипепти́ды[1]) — высокомолекулярные органические вещества, состоящие из альфа-аминокислот, соединённых в цепочку пептидной связью . В живых организмах аминокислотный состав белков определяется генетическим кодом, при синтезе в большинстве случаев используется 20 стандартных аминокислот. Множество их комбинаций создают молекулы белков с большим разнообразием свойств. Кроме того, аминокислотные остатки в составе белка часто подвергаютсяпосттрансляционным модификациям, которые могут возникать и до того, как белок начинает выполнять свою функцию, и во время его «работы» в клетке. Часто в живых организмах несколько молекул разных белков образуют сложные комплексы, например, фотосинтетический комплекс.
Кристаллы различных белков, выращенные на космической станции «Мир» и во время полётовшаттлов НАСА. Высокоочищенные белки при низкой температуре образуют кристаллы, которые используют для изученияпространственной структуры данного белка
Функции белков в клетках живых организмов более разнообразны, чем функции других биополимеров — полисахаридов и ДНК. Так, белки-ферменты катализируют протекание биохимических реакций и играют важную роль в обмене веществ. Некоторые белки выполняют структурную или механическую функцию, образуя цитоскелет, поддерживающий форму клеток. Также белки играют ключевую роль в сигнальных системах клеток, при иммунном ответе и в клеточном цикле.
Белки — важная часть питания животных и человека (основные источники: мясо, птица, рыба, молоко, орехи, бобовые, зерновые; в меньшей степени: овощи, фрукты, ягоды и грибы), поскольку в их организмах не могут синтезироваться все необходимые аминокислоты и часть должна поступать с белковой пищей. В процессе пищеварения ферменты разрушают потреблённые белки до аминокислот, которые используются для биосинтеза собственных белков организма или подвергаются дальнейшему распаду для получения энергии.
Определение аминокислотной последовательности первого белка — инсулина — методом секвенирования белков принеслоФредерику Сенгеру Нобелевскую премию по химии в 1958 году. Первые трёхмерные структуры белков гемоглобина и миоглобинабыли получены методом дифракции рентгеновских лучей, соответственно, Максом Перуцем и Джоном Кендрю в конце 1950-х годов[2][3], за что в 1962 году они получили Нобелевскую премию по химии.
25.1
Основной метод синтеза сложных эфиров заключается в этери - фикации карбоновых кислот спиртами:
RCOOH + R'OH 7—* RC00R' + H20
Скорость образования эфира в значительной степени зависит от строения карбоновой кислоты и спирта. Влияние строения кар - боновой кислоты и спирта на скорость этерификации, а также кинетика этерификации и гидролиза изучены довольно подробно [4-8].
Этерификация является автокаталитическим процессом, т. е. сама карбоновая кислота служит катализатором. Однако при проведении этерификации в отсутствие катализатора часто не достигается необходимой глубины превращения исходных соединений. Кроме того, требуется применение растворителя, большой избыток соответствующего оксисоединения и значительные энергетические затраты.
В качестве катализаторов реакции этерификации карбоновых кислот оксисоединениями используются разнообразные химические соединения.
Традиционными катализаторами этерификации карбоновых кислот оксисоединениями. служат минеральные кислоты: серная, соляная, ортофосфорная, борная [9, 10]. Серная кислота была первым катализатором, примененным для ускорения реакции этерификации. До настоящего времени она остается одним из самых высокоактивных и широко используемых катализаторов промышленного производства сложных эфиров [11—13].
Достоинством серной кислоты как катализатора является значительная скорость этерификации при сравнительно невысоких температурах (80—150°С). К недостаткам серной кислоты как катализатора следует отнести возможность дегидратации спиртов до олефинов, сульфирование ненасыщенных соединений, присутствующих в исходных спиртах и образующихся в результате побочных реакций. Не исключается возможность осмолення органических соединений, а также образование сложных эфиров сульфокислот, что приводит к снижению цветостабильности пластификатора. Для удаления катализатора из сложного эфира-сырца необходимо проводить нейтрализацию щелочным агентом и ряд водных промывок.
При этерификации карбоновых кислот оксисоединениями, катализируемой неорганическими кислородсодержащими и органическими кислотами трех - и пятивалентного фосфора (фосфорнова- тистой, фосфористой, фосфорной) последние не выделяются из реакционной массы (так как они малолетучи), не вызывают осмолення органических веществ и позволяют получить слабоокрашен - ный пластификатор. Бесцветные сложные эфиры образуются при использовании небольших количеств катализатора в сочетании с такими активными адсорбентами как силигокель и диатомит. Однако эти катализаторы имеют меньшую активность, чем серная кислота, и в основном им присущи те же недостатки.
В патентной литературе указывается на применение соединений большинства элементов, встречающихся в природе [9, 10].
Гидролиз
Для гидролиза сложных эфиров и всех остальных производных кислот необходим кислый или щелочной катализ. При кислом гидролизе получают карбоновые кислоты и спирты (реакция обратная этерификации), при щелочном гидролизе образуются соли карбоновых кислот и спирты.
Кислый гидролиз сложных эфиров:

Механизм SN, нуклеофил - H2 O, происходит замещение алкоксигруппы на гидроксил.
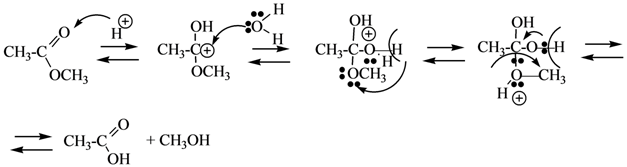
Щелочной гидролиз сложных эфиров: реакция идет в два этапа с 2-мя молями основания, образовавшаяся кислота превращается в соль.
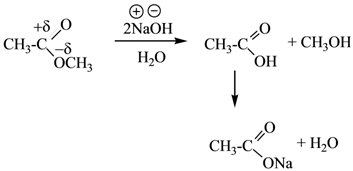
Механизм SN, Nu = −OH
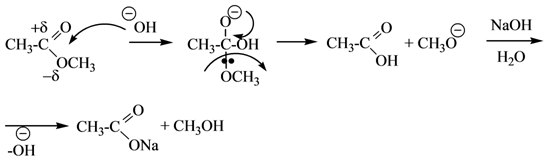
25.2
Образование солеобразных соединений Амиды представляют собой нейтральные вещества, так как основные свойства аммиака ослаблены замещением в нем атома водорода кислотным остатком. Поэтому группа NH2 в амидах в отличие от аминов лишь с трудом образует ониевый катион. Все же с сильными кислотами амиды дают соли, например [CH3CONH3]Cl, легко разлагающиеся водой. С другой стороны, водород группы NH2 в амидах легче, чем ваммиаке и в аминах, замещается на металлы. Ацетамид, например, легко растворяет окись ртути, образуя соединение (CH3CONH)2Hg.
Возможно, однако, что при образовании металлических производных происходит изомеризация амида и получающееся соединение имеет изомерное (таутомерное) строение соли имидокислоты

т. е. здесь имеет место аналогия с солями синильной кислоты.
2. Действие азотистой кислоты С азотистой кислотой амиды реагируют, подобно первичным аминам, с образованием карбоновых кислот и выделением азота:
![]()
3. Омыление При кипячении с минеральными кислотами и щелочами амиды присоединяют воду, образуякарбоновую кислоту и аммиак:
![]()
4. Действие галоидных алкилов. При действии галоидных алкилов на амиды или их металлические производные получаются N-замещенные амиды:
![]()
5. Действие пятихлористого фосфора. При действии пятихлористого фосфора на амиды получаются хлорамиды

легко распадающиеся на соляную кислоту и имидхлориды

Последние с аммиаком могут давать соли амидинов;

6. Превращение в амины. Энергичным восстановлением амидов могут быть получены первичные амины с тем же числом атомов углерода:
![]()
7. Реакция Гофмана. При действии на амиды гипогалогенита или брома и щелочи образуются амины, а углеродныйатом карбонильной группы отщепляется в виде СО2 (А. Гофман). Ход реакции можно представить так:

В учебных руководствах до сих пор еще часто встречается другое толкование механизма этой реакции:

Однако такой ход реакции менее правдоподобен, так как образование осколка

с атомом азота, несущим две свободные электронные пары, мало вероятно.
Против этого механизма говорит, в частности, тот факт, что если радикал R оптически деятельный, то он не рацемизуется в результате реакции. Между тем даже мимолетное существование свободного радикала R–: привело бы к потере оптической деятельности.
26.1
Химические свойства. Нитрогруппа - одна из наиб. сильных электроноакцепторных групп и способна эффективно делокализовать отрицат. заряд. В ароматич. соед. в результате индукционного и особенно мезомерного эффектовона влияет на распределение электронной плотности: ядро приобретает частичный положит. заряд, к-рый локализован гл. обр. в орто- и пара-положениях; константы Гаммета для группы NO2 sм 0,71, sn 0,778, s+n 0,740, s-n1,25. Т. обр., введение группы NO2 резко увеличивает реакц. способность орг. соед. по отношению к нуклеоф.реагентам и затрудняет р-ции с электроф. реагентами. Это определяет широкое применение нитросоединений в орг. синтезе: группу NO2 вводят в нужное положение молекулы орг. соед., осуществляют разл. р-ции, связанные, как правило, с изменением углеродного скелета, и затем трансформируют в др. ф-цию или удаляют. В ароматич. ряду часто используют и более короткую схему: нитрование-трансформация группы NO2.
Образование нитроновых к-т в ряду ароматических нитросоединений связано с изомеризацией бензольного кольца в хиноидную форму; напр., нитробензол образует с конц. H2SO4 окрашенный солеобразный продукт ф-лы I, о-нитротолуол проявляет фотохромизм в результате внутримол. переноса протона с образованием ярко-синего О-производного:

При действии оснований на первичные и вторичные нитросоединения образуются соли нитросоединений; амбидентные анионы солей в р-циях с электрофилами способны давать как О-, так и С-производ-ные. Так, приалкилировании солей нитросоединений алкилгалогенидами, триалкилхлорсиланами или R3O+BF-4 образуются продукты О-алкилирования. Последние м.б. получены также при действии диазометана либо N,О-бис-(триметилсилил)аце-тамида на нитроалканы с рКа < 3 или нитроновые к-ты, напр.:
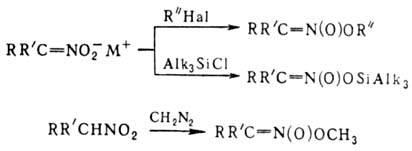
Ациклич. алкиловые эфиры нитроновых к-т термически нестабильны и распадаются по внутримол. механизму:
![]() ;
эту
;
эту
Р-ц и и с р а з р ы в о м с в я з и С—N. Первичные и вторичные нитросоединения при нагр. с минер. к-тами в присут. спиртового или водного р-ра щелочи образуют карбонильные соед. (см. Нефа реакция). Р-ция проходит через промежут. образование нитроновых к-т:

В качестве исходных соед. можно использовать силиловые нитроновые эфиры. Действие сильных к-т на алифатические нитросоединения может приводить к гидроксамовым к-там, напр.:
Известно много методов восстановления нитросоединений до аминов. Широко используют железные опилки, Sn и Zn в присут. к-т; при каталитич. гидрировании в качестве катализаторов используют Ni-Ренея, Pd/C или Pd/PbCO3 и др. Алифатические нитросоединения легко восстанавливаются до аминов LiAlH4 и NaBH4 в присут. Pd, амальгамамиNa и Аl, при нагр. с гидразином над Pd/C; для ароматических нитросоединений иногда применяют ТlСl3, СrСl2 и SnCl2, ароматич. поли-нитросоединения избирательно восстанавливаются до нитраминов гидросульфидом Na в СН3ОН. Существуют способы избират. восстановления группы NO2 в полифункциональных нитросоединениях без затрагивания др. ф-ций.
При действии Р(III) на ароматические нитросоединения происходит последоват. дезоксигенирование группы NO2 с образованием высокореакционноспособных нитренов. Р-цию используют для синтеза конденсир. гетероциклов, напр.:

Р-ц и и с с о х р а н е н и е м г р у п п ы NO2. Алифатические нитросоединения, содержащие a-Н-атом, легко алкилируются и ацилируются с образованием, как правило, О-производных. Однако взаи-мод. дилитиевых солейпервичных нитросоединений с алкилгалогенидами, ангидридами или галогенангидридами карбоновых к-т приводит к продуктам С-алкилирования или С-ацилирования, напр.:

Известны примеры внутримол. С-алкилирования, напр.:
![]()
Первичные и вторичные нитросоединения реагируют с алифатич. аминами и СН2О с образованием р-аминопроизводных (р-ция Манниха); в р-ции можно использовать предварительно полученные метилольные производные нитросоединений или аминосоед.:
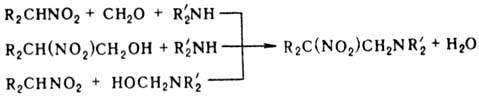
Нитрометан и нитроэтан могут конденсироваться с двумя молекулами метилоламина, а высшие нитроалканы- только с одной. При определенных соотношениях реагентов р-ция может приводить к гетероциклич. соед., напр.: при взаимод. первичного нитроалкана с двумя эквивалентами первичного амина и избытком формальдегидаобразуются соед. ф-лы V, если реагенты берут в соотношении 1:1:3-соед. ф-лы VI.

Ароматические нитросоединения легко вступают в р-ции нуклеоф. замещения и значительно труднее-в р-ции электроф. замещения; при этом нуклеофил на правляется в орто- и пора-поло жения, а электрофил-в мета-положение к группе NO2. Константа скорости электроф. нитрования нитробензола на 5-7 порядков меньше, чембензола; при этом образуется м-динитробензол.
При карбоксилировании первичных нитроалканов действием CH3OMgOCOOCH3 образуются a-нитрокарбоновые к-ты или их эфиры.
При обработке солей моно-нитросоединений C(NO2)4., нитритами Ag или щелочных металлов либо при действиинитритов на a-гало-геннитроалканы в щелочной среде (р-ция Тер Меера) образуются гем-динитросоединения.Электролиз a-галоген-нитроалканов в апротонных р-рителях, а также обработка нитросоединений Сl2 в щелочной среде или электроокисление солей нитросоединений приводят к виц-динитросоединениям:

Нитрогруппа не оказывает существ. влияния на свободно-радикальное алкилирование или арилирование ароматич. соед.; р-ция приводит в осн. к орто- и пара-замещенным продуктам.
Для восстановления нитросоединений без затрагивания группы NO2 применяют NaBH4, LiAlH4 при низких т-рах или р-р дибора-на в ТГФ, напр.:
![]()
Ароматич. ди- и три-нитросоединения, в частности 1,3,5-тринитробен-зол, образуют устойчивые ярко окрашенные кристаллич. мол. комплексы с ароматич. соед.-донорами электронов (аминами, фенолами и др.). Комплексы с пикриновой к-той используют для выделения и очистки ароматич. углеводородов. Взаимод. ди- и тринитробензоловс сильными основаниями (НО-, RO-, N-3, RSO-2, CN-, алифатич. аминами) приводит к образованию комплексов Майзен-хаймера, к-рые выделяют в виде окрашенных солей щелочных металлов.
27.1
В качестве окислителей для этих реакций пригодны хромовая или азотная кислота, хромовая смесь, двуокись марганца или двуокись селена.
При окислении хромовой кислотой спирт нуклеофильно присоединяется к хромовой кислоте, при этом отщепляется вода и образуется эфир хромовой кислоты (это первая стадия реакции, она аналогична образованию сложных эфиров карбоновых кислот, ср. разд. Д,7.1.5.1). Во второй стадии, идущей, вероятно, через циклическое переходное состояние, a-водород спирта переходит к остатку хромата, причем металл из шестивалентного состояния переходит в четырехвалентное:
|
|
|
|
(Г.6.18) |

Четырехвалентный хром далее восстанавливается спиртом до трехвалентного состояния, так что окончательный результат можно записать с помощью следующего уравнения:
|
|
|
|
(Г.6.19) |
Для замещенных в ядре производных 1-фенилэтанола установлена следующая зависимость окисляемости от заместителя:
|
|
|
n-CH3O > п-трет-С4H9 > п-СН3 > п-Сl > п-NO2 |
(Г.6.20) |
При окислении первичных спиртов образующийся альдегид должен быть защищен от дальнейшего окисления в карбоновую кислоту. Можно, например, постоянно отгонять альдегид из реакционной смеси: это вполне осуществимо, так как температура кипения альдегида обычно ниже, чем температура кипения соответствующего спирта. Все же выход альдегидов при окислении бихроматом редко превышает 60%. Примечательно, что при надлежащем проведении реакции кратные углерод-углеродные связи почти не затрагиваются.
Альдегиды образуются также при нагревании спиртов с водным нейтральным раствором бихромата, однако хорошие выходы при этом дают лишь бензиловые спирты.
Более высокие выходы альдегидов можно получить при окислении первичных спиртов трет-бутилхроматом (в петролейном эфире, бензоле или четыреххлористом углероде) или двуокисью марганца (в ацетоне, петролейном эфире, четыреххлористом углероде или разбавленной серной кислоте). Эти реагенты позволяют с хорошими выходами получать также ненасыщенные и ароматические альдегиды.
Окисление вторичных спиртов до кетонов осуществляется еще легче, чем окисление первичных спиртов. Выходы здесь выше, так как, во-первых, реакционная способность вторичных спиртов выше, чем первичных, а во-вторых, образующиеся кетоны гораздо устойчивее к окислению по сравнению с альдегидами. В ряду стероидов и терпенов хорошо зарекомендовало себя окисление вторичных спиртов комплексом хромовой кислоты с пиридином, а также хромовым ангидридом в диметилформамиде. Хорошим окислителем является также хромовый ангидрид в ацетоне; с его помощью можно окислять ненасыщенные вторичные спирты, не затрагивая кратную углерод-углеродную связь.
Новым методом, пригодным и для пространственно затрудненных спиртов, является окисление диметилсульфоксидом в уксусном ангидриде.
Согласно приводимой ниже методике, реакция ведется в двухфазной системе. Образовавшиеся кетоны извлекаются органическим растворителем и таким образом предохраняются от дальнейшего окисления.
27.2
Дисахариды – углеводы, молекулы которых состоят из двух остатков моносахаридов, которые соединены друг с другом за счет взаимодействия двух гидроксильных групп.
В процессе образования молекулы дисахарида происходит отщепление одной молекулы воды:
![]()
или для сахарозы:
![]()
Поэтому молекулярная формула дисахаридов С12H22O11.
Образование сахарозы происходит в клетках растений под воздействием ферментов. Но химики нашли способ осуществления многих реакций, являющихся частью процессов, которые происходят в живой природе. В 1953 году французский химик Р. Лемье впервые осуществил синтез сахарозы, названный современниками «покорением Эвереста органической химии».
В промышленности сахароза получается из сока сахарного тростника (содержание 14-16%), сахарной свеклы (16-21%), а также некоторых других растений, таких как канадский клен или земляная груша.
Всем известно, что сахароза представляет из себя кристаллическое вещество, которое имеет сладкий вкус и хорошо растворимо в воде.
Сок сахарного тростника содержит углевод сахароза, привычно называемый нами сахаром.
Имя немецкого химика и металлурга А. Маргграфа тесно связано с производством сахара из свеклы. Он был одним из первых исследователей, применивших в своих химических исследованиях микроскоп, при помощи которого им были обнаружены кристаллы сахара в свекольном соке в 1747 году.
Лактоза – кристаллический молочный сахар, была получена из молока млекопитающих еще в XVII в. Лактоза является менее сладким дисахаридом, нежели сахароза.
Теперь ознакомимся с углеводами, имеющими более сложное строение – полисахаридами.
Полисахариды – высокомолекулярные углеводы, молекулы которых состоят из множества моносахаридов.
В упрощенном виде общая схема может быть представлена так:
![]()
Теперь сравним строение и свойства крахмала и целлюлозы – важнейших представителей полисахаридов.
Структурное звено полимерных цепей этих полисахаридов, формула которых (С6H10O5)n, – это остатки глюкозы. Для того, чтобы записать состав структурного звена (С6H10O5), нужно отнять молекулу воды из формулы глюкозы.
Целлюлоза и крахмал имеют растительное происхождение. Они образуются из молекул глюкозы в результате поликонденсации.
Уравнение реакции поликонденсации, а также обратного ей процесса гидролиза для полисахаридов условно можно записать следующим образом:
![]()
Молекулы крахмала могут иметь как линейный, так и разветвленный тип строения, молекулы целлюлозы – только линейный.
При взаимодействии с йодом крахмал, в отличие от целлюлозы, дает синее окрашивание. Различные функции эти полисахариды имеют и в растительной клетке. Крахмал служит запасным питательным веществом, целлюлоза выполняет структурную, строительную функцию. Стенки растительных клеток построены из целлюлозы.
29.1
КАННИЦЦАРО РЕАКЦИЯ,
окислит.-восстановит. диспропорционирование альдегидов под
действием щелочи с
образованием первичных спиртов и
карбоновых к-т, напр.:
![]() Альдегид обрабатывают
конц. водным или водно-спиртовым
р-ром щелочи при
охлаждении или слабом нагревании.Катализаторы -
разл. металлы (напр.,
Ag, Ni, Co, Сu) и их оксиды.
В р-цию вступают альдегиды,
не содержащие атомН
в a-положении
к карбонильной группе. В противном
случае предпочтительней идет не
Канниццаро реакция,
аальдольная
конденсация.
Электроноакцепторные заместители в
кольце ароматич. альдегидов ускоряют
процесс, а электронодонорные
замедляют. Бензальдегиды с
заместителями в орто-положениях в
Канниццаро реакцию не
вступают; о- и п-гидроксибензальдегиды
реагируют только в присут. Ag. Р-цию
с использованием двух разл.альдегидов (т.
наз. перекрестная Канниццаро реакция)
применяют гл. обр. для получения с большим
выходом первичных спиртов из
ароматич. альдегидов.
В качестве восстановителя при
этом обычно выступает формальдегид:
Альдегид обрабатывают
конц. водным или водно-спиртовым
р-ром щелочи при
охлаждении или слабом нагревании.Катализаторы -
разл. металлы (напр.,
Ag, Ni, Co, Сu) и их оксиды.
В р-цию вступают альдегиды,
не содержащие атомН
в a-положении
к карбонильной группе. В противном
случае предпочтительней идет не
Канниццаро реакция,
аальдольная
конденсация.
Электроноакцепторные заместители в
кольце ароматич. альдегидов ускоряют
процесс, а электронодонорные
замедляют. Бензальдегиды с
заместителями в орто-положениях в
Канниццаро реакцию не
вступают; о- и п-гидроксибензальдегиды
реагируют только в присут. Ag. Р-цию
с использованием двух разл.альдегидов (т.
наз. перекрестная Канниццаро реакция)
применяют гл. обр. для получения с большим
выходом первичных спиртов из
ароматич. альдегидов.
В качестве восстановителя при
этом обычно выступает формальдегид:
АrСНО + СН2О : АrСН2ОН + НСООН
При синтезе
полигидроксиметилированных
соед. формальдегид на
первой стадии участвует в альдольной
конденсации,
а затем в качестве восстановителя в
перекрестной Канниццаро реакции:
 Предполагаемый
механизм Канниццаро реакции в
гомог. среде включает стадию гидридного
переноса
Предполагаемый
механизм Канниццаро реакции в
гомог. среде включает стадию гидридного
переноса
 Для
ароматич. альдегидов не
исключена возможность участия в
Канниццаро реакции анион-радикалов,
образующихся в результате одноэлектронного
переноса. Р-ция,
подобная Канниццаро реакции,
осуществляется при
внутримол. диспропорционировании a-кетоальдегидов
в присут. щелочей (перегруппировка
Канниццаро):
Для
ароматич. альдегидов не
исключена возможность участия в
Канниццаро реакции анион-радикалов,
образующихся в результате одноэлектронного
переноса. Р-ция,
подобная Канниццаро реакции,
осуществляется при
внутримол. диспропорционировании a-кетоальдегидов
в присут. щелочей (перегруппировка
Канниццаро):
 Канниццаро реакцию применяют
для пром. синтеза пентаэритрита,
препаративного получения спиртов,
карбоновых к-т и др. Р-ция
открыта С. Канниццаро в 1853.
Канниццаро реакцию применяют
для пром. синтеза пентаэритрита,
препаративного получения спиртов,
карбоновых к-т и др. Р-ция
открыта С. Канниццаро в 1853.
29.2
Пиррол, фуран и тиофен являются пятичленными гетероциклическими соединениями с одним гетероатомом.

Нумерация атомов в составе гетероцикла начинается с гетероатома и идет против часовой стрелки. Положения 2- и 5-называют a-положениями, 3- и 4- – b-положениями.
По формальным признакам эти соединения относятся к ароматическим, так как они представляют собой сопряженные циклические p-системы, в состав которых входит 6p электронов – 4 электрона диеновой системы – и пара электронов гетероатома. Цикл является практически плоским, из чего следует, что состояние гибридизации гетероатома близко к sp2.
Ниже представлены резонансные структуры, иллюстрирующие делокализацию электронов гетероатома по гетероциклическому кольцу на примере фурана.

Приведенные резонансные структуры показывают, что гетероатом (в данном случае атома кислорода) в результате мезомерного взаимодействия с диеновой π-системой передает электронную плотность в кольцо, вследствие чего на атомах углерода в составе гетероцикла возникает некоторый отрицательный заряд, а на атоме кислорода, соответственно, положительный заряд. Атом кислорода, разумеется, кроме положительного мезомерного эффекта проявляет и отрицательный индуктивный эффект. Однако его проявление в свойствах рассматриваемых соединений менее выражено, в связи с чем пятичленные гетероциклы с одним гетероатомом относят к p-избыточным ароматическим гетероциклическим соединениям. Резонанс приводит к некоторой выравненности длин связей в составе гетероцикла, что также говорит об определенной ароматичности системы.