

Автоматический газовый хроматографический анализ
..pdfпостороннего компонента (метод внутреннего стандарта) или через площадь пика компонента сравнения (метод стандартной добавки).
5.4.1. Метод абсолютной калибровки
Метод основан на использовании зависимости между количеством компонента в пробе и параметрами пика. Эту зависимость определяют экспериментально, хроматографируя искусственные смеси, и выражают либо в виде графика «Параметр пика — количество (концентрация)» (рис. 34), либо аналитически:
Ci |
km |
Si |
100, |
масс. % |
(81) |
||
mn |
|||||||
|
ai |
|
|
|
|
|
|
или |
kV |
Si |
|
|
|
||
Ci |
100, |
об. %, |
(82) |
||||
V |
|
|
|||||
|
ai |
|
|
|
|
|
|
n
где mn и Vn — соответственно масса и объем введенной пробы.
Калибровка проводится как по определяемым, так и по стандартным соединениям. В последнем случае должны быть известны отношения массовых или молярных чувствительностей детектора к этим соединениям. Калибровку можно проводить, используя либо одинаковые по объему пробы, либо различные объемы постоянной концентрации. Первый способ обычно используют при анализе жидких проб, а второй – при анализе газообразных.
Метод абсолютной калибровки используется при работе как с линейными детекторами, так и с нелинейными детекторами и при искажении параметра пика вследствие перегрузки колонки. При использовании этого метода требуется разделение только интересующих компонентов, поэтому он может применяться и при обратной продувке колонки, и при отсутствии отклика детектора к некоторым соединениям.
Метод абсолютной калибровки требует соблюдения полной идентичности условий хроматографического процесса при калибровке прибора и анализе исследуемой смеси. Необходимую информацию о работе прибора (газового хроматографа) может дать анализ стандартной смеси.
Точность результатов, получаемых этим методом, зависит от точности дозирования пробы. Этот метод рекомендуется использовать при введении пробы газовым краном-дозатором. Метод используется при контроле и регулировании технологических процессов с помощью промышленных (потоковых) хроматографов. Метод является основным при анализе примесей.
111
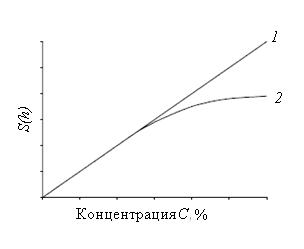
Рис. 35. Зависимость параметра пика от количества (концентрации): 1 — прямолинейная зависимость; 2 –– криволинейная зависимость
5.4.2. Метод внутренней нормализации
Этот метод осуществляется в виде нескольких вариантов. В методе простой нормализации сумма площадей всех пиков принимается за 100 % и концентрация любого компонента пробы рассчитывается как относительная площадь пика:
Ci |
Si |
100. |
(83) |
n |
Si
i 1
Необходимым условием применения этого метода является регистрация всех компонентов пробы и одинаковая чувствительность детектора к разным веществам. Для большинства детекторов это, в общем, справедливо, если анализируется смесь родственных соединений, молекулярные массы которых значительно не различаются или все компоненты пробы имеют большие молекулярные массы. Например, не требуется калибровка при анализе смеси циклогексана и бензола или при анализе изомеров ксилола. Этот вариант метода имеет ограниченное применение. В большинстве случаев приходится учитывать разный отклик детектора к различным веществам пробы с помощью калибровочных коэффициентов, зависящих от свойств вещества, способа детектирования, а также от конструкции детектора.
В основном варианте метода расчет проводится с учетом калибровочных коэффициентов:
Ci |
Si Ki |
100, |
(84) |
n |
|||
|
Si Ki |
|
|
|
i 1 |
|
|
где Сi — концентрация компонента i, %; |
|
||
Si — площадь соответствующего пика; |
|
||
|
|
n |
|
Ki — калибровочный коэффициент; Si Ki |
— сумма произведений |
||
|
|
i 1 |
|
площадей пиков на относительные поправочные коэффициенты для всех пиков хроматограммы.
112

Достоинства метода внутренней нормализации состоят в том, что он не требует воспроизводимого ввода пробы по величине и тождественности условий анализа. Расчеты проводятся с использованием относительных калибровочных коэффициентов, которые малочувствительны к небольшим изменениям в условиях проведения эксперимента.
Недостатки метода заключаются в трудности разделения всех компонентов сложных смесей, необходимости их идентификации и трудоемкости определения калибровочных коэффициентов, хотя некоторые компоненты могут и не представлять аналитического интереса. Недостатком метода является зависимость точности определения одного компонента от точности определения остальных компонентов, присутствующих в смеси. Ошибки в определении параметров пика или калибровочного коэффициента какого-либо одного компонента приводят к неверным результатам для всех компонентов пробы.
Метод используется в основном для рутинных анализов малокомпонентных смесей и для приближенных расчетов.
5.4.3. Метод внутреннего стандарта
Метод заключается в том, что к навеске анализируемого вещества добавляется известное количество внутреннего стандарта — постороннего соединения, дающего на хроматограмме хорошо разрешенный пик. Концентрация определяемого компонента в анализируемом веществе рассчитывается по формуле:
Ci |
Si Ki M ст |
100, масс. % |
(85) |
|
|||
|
Sст M п |
|
|
где Sст — площадь пика стандарта;
Mст — масса добавленного внутреннего стандарта;
M п — масса пробы анализируемой смеси, к которой добавлено определенное количество внутреннего стандарта.
Приведенное уравнение может служить основой графического варианта рассматриваемого метода. В самом деле, это уравнение прямой, построенной в координатах
Ci Si M ст 100.
Sст M п
Если Мcт/Mп остается постоянным, тогда получают калибровочные графики в координатах:
Ci Si .
Sст
Рассмотрим подробнее требования к внутреннему стандарту. Внутренний стандарт должен:
1)иметь хорошо разрешенный пик, расположенный на хроматограмме рядом с пиками определяемых компонентов;
2)иметь летучесть, близкую определяемым компонентам;
3)отсутствовать в анализируемой смеси;
113
4)хорошо растворяться в анализируемой смеси, не реагировать с другими компонентами пробы;
5)добавляться в количестве, соизмеримом с анализируемыми компонентами.
Выполнение первого условия дает возможность точного измерения параметра пика, уменьшает влияние изменения чувствительности детектора из-за колебаний рабочих условий (расход газа-носителя, температура колонки
ит.д.); второго — уменьшает ошибки, вызванные фракционированием пробы при вводе; третьего и четвертого — обеспечивает точное значение массы внутреннего стандарта; пятого — уменьшает влияние нелинейности детектора к различным количествам пробы.
Достоинства метода внутреннего стандарта состоят в том, что он не требует воспроизводимого ввода пробы по величине; малая зависимость результатов измерений от нестабильности работы хроматографа и детектора, т.к. эти факторы в равной мере влияют на определяемое и стандартное соединение. Также, к достоинствам метода относится то, что здесь требуется разделять только анализируемые компоненты и стандарт. Ошибки в измерении параметров пика и калибровочного коэффициента (исключая стандарт) сказываются только на определении содержания соединения, для которого была допущена ошибка.
К недостаткам метода следует отнести трудность выбора, в ряде случаев, соединения-стандарта, удовлетворяющего перечисленным условиям.
Метод внутреннего стандарта применяется в основном при анализе жидкостей, поскольку при анализе газов трудно дозировать определенное количество внутреннего стандарта в газовую смесь.
5.4.4. Метод стандартной добавки
В ряде случаев при анализе сложных смесей выбор внутреннего стандарта вызывает затруднение. Здесь можно воспользоваться методом стандартной добавки. Для определения концентрации этим методом необходимо записать две хроматограммы:
анализируемой смеси;
обогащенной определяемым компонентом исходной смеси (анализируемая смесь + добавка определяемого компонента).
Концентрация в исходной смеси определяемого компонента и веществастандарта выражаются формулами:
Ci |
M ст |
|
|
|
|
|
|
100 |
|
, масс. % |
(86) |
|||
M п |
|
|
Sст2 |
|
|
|
Sст1 |
|||||||
|
|
|
|
Si |
Ki |
|
Si Ki |
|
|
|||||
|
M |
|
|
|
2 |
100 |
|
1 |
|
|
|
|||
Ci |
|
|
|
|
|
|
, |
|
(87) |
|||||
ст |
|
|
|
|
|
|
|
|
||||||
M |
|
S |
ст2 |
|
Si |
|
|
|||||||
|
п |
|
|
|
|
|
1 |
|
1 |
|
|
|||
|
|
|
|
Si |
|
|
Sст1 |
|
|
|
|
|||
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
где Sстl и Sст2 — площадь пиков, принятых за стандарт, соответственно на хроматограмме исходной смеси и после добавления в нее стандарта
114
(компонента, содержащегося в смеси);
Si1 и Si2 – площадь пиков определяемого компонента i на хроматограмме исходной смеси и на хроматограмме смеси, в которую введен стандарт, соответственно;
Ki — относительный (или абсолютный) поправочный коэффициент для компонента i.
К достоинствам метода стандартной добавки следует отнести то, что не требуется знания калибровочных коэффициентов. Недостаток — возрастающая продолжительность проведения анализа.
5.4.5. Экспериментальное определение абсолютных и относительных калибровочных коэффициентов
Абсолютные поправочные коэффициенты ki — коэффициенты пропорциональности между количеством соединения, введенным в
хроматограф, и интенсивностью сигнала детектора. Они зависят от типа и конструкции детектора, природы анализируемого соединения и условий анализа. С введением этих коэффициентов количественный хроматографический параметр (высота, площадь) в условиях эксперимента становится пропорциональным только количеству соответствующего соединения:
ki |
mi |
. |
(88) |
|
|||
|
Si |
|
|
Для определения ki необходимо приготовить смесь строго определенных количеств (по массе или объему) анализируемых компонентов. Содержание компонентов в модельной смеси должно быть по возможности близким к составу анализируемой смеси. Далее проводят хроматографическое разделение определенного объема (или массы) модельной смеси и рассчитывают количество введенного в хроматограф данного компонента смеси по формуле
mi M |
qi |
100, |
(89) |
|
Q |
||||
|
|
|
где mi — количество компонента i, введенное в хроматограф;
М — количество всей смеси, введенной в хроматограф; qi — содержание компонента i в модельной смеси;
Q — общее количество приготовленной модельной смеси
После этого измеряют площади пиков компонентов и по приведенной выше формуле (87) рассчитывают поправочные коэффициенты.
Относительный массовый калибровочный коэффициент fi вычисляют по формуле
fi |
mi Sст |
|
ai Sст |
, |
(90) |
|||||
|
|
|||||||||
|
m |
S |
i |
|
a |
ст |
S |
i |
|
|
|
ст |
|
|
|
|
|
|
|||
где mi и mст — масса введенного в хроматограф компонента i смеси и компонента, принятого за стандарт, соответственно;
Si и Sст — площадь пиков компонента i и компонента, принятого за стандарт;
115
ai и аст — содержание в смеси компонента i и стандарта, выраженное в масс. %.
Относительные мольные поправочные коэффициенты используют в тех случаях, когда конечные результаты расчетов по тем или иным причинам желательно получить в молях или в мольных процентах:
fi mol |
Ni Sст |
|
bi Sст |
, |
(91) |
|||||
|
|
|||||||||
|
N |
ст |
S |
i |
|
b |
S |
i |
|
|
|
|
|
|
ст |
|
|
|
|||
где Ni и Nст — число молей i-го компонента и стандарта, введенного в
хроматограф;
bi и bст — содержание в смеси компонента i и стандарта, выраженное в моль %.
Для экспериментального определения относительных калибровочных коэффициентов необходимо приготовить стандартную смесь и записать несколько хроматограмм этой смеси. Расчет относительных калибровочных коэффициентов производится по приведенным выше формулам.
Относительные калибровочные коэффициенты показывают, во сколько раз площадь пика стандартного соединения больше площади пика определяемого компонента, когда введенные количества (концентрации) компонентов одинаковы, т.е. они учитывают различную чувствительность детектора к различным компонентам.
Зависимость между массовым и мольным поправочными коэффициентами определяются следующим соотношением:
fi fi mol |
M i |
, |
(92) |
|
|||
|
M |
|
|
где Mi — молекулярная масса компонента i;
M — молекулярная масса того компонента, для которого коэффициенты fi и fimol приняты за единицу.
116

6. МЕТОДЫ АВТОМАТИЧЕСКОГО ОБНАРУЖЕНИЯ ПИКОВ В ВЫХОДНОМ СИГНАЛЕ ХРОМАТОГРАФА И КОРРЕКЦИИ БАЗИСНОГО СИГНАЛА
6.1.Фильтрация хроматографического сигнала
Под фильтрацией здесь понимается определенная операция над сигналом, целью которой может быть сглаживание или получение некоторых производных сигнала. Применение фильтра не исключает использования средств подавления или защиты от источников помех (соответствующего заземления, экранирования, применения помехоустойчивых схем), а наоборот, облегчает требования к качеству их работы. Для фильтрации могут применяться аналоговые и цифровые фильтры.
6.1.1. Фильтрация хроматографического сигнала от шумов.
Использование в последнее время для обработки хроматографических сигналов цифровых устройств с развитой памятью способствовало широкому применению цифровых программных фильтров. Выходной эффект такого цифрового фильтра может быть представлен в виде:
m |
|
|
yi* Fk yi k |
(m 0) |
(93) |
k m
где Fk — весовые коэффициенты (веса), характеризующие вклад yi+k в значения фильтрованного сигнала в i-й точке.
Простейшим является фильтр скользящего среднего. В этом случае
Fk 2m1 1.
Симметричная фильтрация такого рода может быть осуществлена только над задержанным сигналом или во вторичном времени, несимметричная — в реальном времени.
Фильтр по уравнению (93) из нерекурсивной (оперирующей только входным сигналом) формы можно представить в рекурсивном виде:
p |
m |
|
yi* G j yi* j Fk yi k |
(m, p 0) . |
|
j 1 |
k m |
|
Например, для фильтра скользящего среднего нерекурсивная форма требует (2m + 1) сложений, а рекурсивная только – трех:
yi* yi* 1 |
1 |
|
(yi m yi m 1). |
(94) |
|
2m 1 |
|||||
|
|
|
|||
В случае экспоненциального сглаживания (чем ближе точка к i-й,
117
тем она значимее) нерекурсивный фильтр [Fk = (1 —γ)K γ] требует i умножений:
yi* γ i |
(1 γ) j yi j ), |
(95) |
j0
арекурсивный — только двух (при реализации в оперативной памяти необходимо выделить всего одно слово):
yi* (1 γ)yi* 1 γyi . |
(96) |
Постоянная у управляет степенью сглаживания (при γ = 1 сглаживания
нет).
Воздействия на случайную и детерминированную часть сигнала для линейного фильтра независимы. Для сохранения постоянного сигнала неизменным необходимо провести нормирование весов:
m
Fk 1.
k m
При некоторых условиях фильтрация эквивалентна дифференцированию. Эти условия имеют вид:
m |
m |
ti k i Fk 0 |
t n k n Fk n!, |
k m |
k m |
где i = 1, ..., п — 1; n — порядок производной.
Примером линейного дифференцирующего фильтра для оценки первой производной сигнала может служить фильтр
* |
|
1 |
m |
|
y' i |
|
|
kyi k |
(97) |
m |
||||
|
|
k 2 |
k m |
|
k m
или в рекурсивной форме (для больших т):
y' * y' * |
|
1 |
2m 1 y* my |
i m 1 |
m 1 y |
i m |
, |
(98) |
||
m |
||||||||||
i |
i 1 |
|
i |
|
|
|
||||
|
|
|
k 2 |
|
|
|
|
|
|
|
k m
где yi* находится по (94).
В ряде случаев процедура фильтрации по (93) эквивалентна аппроксимации по методу наименьших квадратов (МНК) сигнала некоторым полиномом степени п. Ординате средней точки выборки присваивается значение полинома в этой точке с коэффициентами, найденными по значениям сигнала в выборке, а затем выборка сдвигается и процедура повторяется.
Рассмотрим искажение сигнала и снижение шума при фильтрации, а
118
также ее влияние на оценки параметров пиков. Наиболее сильные искажения вносят несимметричные фильтры, в частности аналоговые. На рис. 36 показаны искажения гауссова пика после прохождения аналогового RC- фильтра первого порядка.
Рис. 36. Искажения гауссова пика при его прохождении через фильтр первого порядка с постоянной времени Т1 равной: 1 – 0; 2 – 0,6µ; 3 – 1,2µ; 4 –
1,8µ; 5 – 2,4µ; 6 – 3,0µ; 7 – 3,6µ; 8 – 4,2µ; 9 – 4,8µ
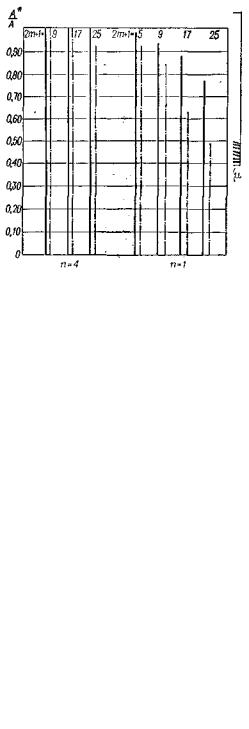
Искажения, вносимые симметричными полиномиальными фильтрами, значительно меньше и возрастают с увеличением асимметрии пика и числа точек в выборке т (рис. 37). Увеличение шага квантования t также ведет к росту искажений.
Для их уменьшения необходимо использовать фильтры с малыми т, но увеличивать п, хотя при этом ухудшается подавление шумов.
Рис. 36. Искажение высоты пика (отношение амплитуд пика до и после (А*) фильтрации) в зависимости от ширины полиномиального фильтра для пиков гауссовой формы (µ* = 10) и асимметричного пика (свертка гауссовой
кривой с экспонентой – штрихпунктир)
119
Изменение t и т приводит к изменению отношения ширины «окна» фильтра ωф и сигнала ωс:
|
ωф |
. |
(99) |
|
|||
|
ωс |
|
|
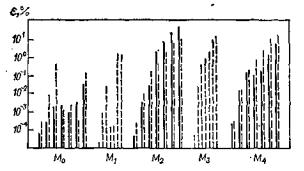
Из рис. 38, на котором приведены графики, характеризующие влияние фильтрации на моменты пиков, следует, что наименее подвержен искажениям нулевой момент (площадь) пика: только при m = 12 погрешности достигают заметных значений и возрастают с асимметрией. Ошибки в вычислении первого момента возрастают лишь при больших ωф.
Рис. 37. Влияние фильтрации n-го порядка на относительную погрешность ε оценки моментов пиков для гауссова пика (µ* = 10) и асимметричного пика (пунктир). Диаграммы для каждого момента соответствуют следующим параметрам: n = 4, (2m + 1) = 9, 17, 25 и n = 2, (2m + 1) = 5, 9, 17, 25
Значительно существеннее искажения, вносимые фильтрацией в оценки моментов более высоких порядков (поэтому значимые результаты могут быть получены только для низкошумящих хроматографических систем с большими отношениями сигнал/шум q). Искажения симметрии пика тем меньше, чем ниже значения m и t и выше порядок фильтрации n.
Повторная фильтрация, как указывалось, сужает полосу пропускания фильтра, при этом искажения возрастают с увеличением числа повторов (прогонов) и я уменьшением n.
Качество передачи гауссова пика в зависимости от величины φ (параметр) и числа повторных фильтраций (n = 2) можно оценить по рис. 39 Различная ширина хроматографических пиков затрудняет использование кривых рис. 39 для коррекции искажений (для каждого пика нужен свой коэффициент).
120