

биохимия атеросклероза
.pdfTLR4
CD36
PTK
Proteasomal degradation
IL-1Rα
IL-1β
IL-6
TNF-α/β
IFN-γ/β
FIGURE 5.1. Hypothetical model of CD36–TLR4 interactions and NF-κB activation. NF-κB is an evolutionarily conserved transcription factor required for many aspects of the immune and inflammatory response. In atherosclerosis, NF-κB coordinates the expression of a wide array of inflammatory genes (reviewed in [100]) and is thus central to the pathology of this disease. The activity of NF-κB is tightly regulated by I-κB inhibitory proteins, which bind to NF-κB, sequestering it in the cytoplasm. When the NF-κB pathway is activated, I-κBs are phosphorylated by kinases (IKKs) leading to the dissociation of the NF-κB:I-κB complex and NF-κB translocation to the nucleus. In the nucleus, NF-κB binds to the promoter of target genes and stimulates gene expression. An important regulator of the NF-κB pathway is the toll-like receptor (TLR) family of pattern recognition receptors. TLRs are crucial to the innate host response to infectious agents such as bacteria due to their ability to bind endotoxin/LPS. The growing body of data linking such pathogens with atherosclerosis has cast a new light on the role of TLRs in lesion development and progression. In particular, the TLR4 pathway has garnered much attention (reviewed in [101]). Ligand engagement of TLR4 triggers the recruitment of intracellular adapter proteins such as MyD88, which results in the activation of numerous kinases, phosphorylation of I-κB and thus elevated NF-κB activity. We hypothesize that CD36–TLR4 aggregation results in the recruitment of CD36associated protein tyrosine kinases (PTKs), which further enhance IKK and NF-κB activity. Potential sites of PTK activity are indicated by dashed arrows as are some of the genes which may be affected by CD36 expression levels [45].
Chapter 5. The Role of SRs in Signaling, Inflammation, and Atherosclerosis |
79 |
and c-Src, has also been suggested [48]. Together, the data strongly argue for a functional effect of CD36 on NF-κB activation, perhaps via intracellular kinases associated with the C-terminal cytoplasmic tail of CD36.
Physical interactions between CD36 and a major regulatory protein in the NF-κB pathway have also been detected. As noted in Fig. 5.1, the toll-like receptor (TLR)-4 is an important mediator of NF-κB and has been well characterized as a major receptor for LPS, a glycolipid found on the outer membrane of gram-negative bacteria [49–51]. Similar to other immunoreceptors, the first step in TLR4 signaling is ligand-dependent receptor aggregation, which initiates signaling cascades and thus leads to the appropriate inflammatory response. CD36 appears to participate in this receptor aggregation. In human monocytes, stimulation with LPS induced the aggregation of TLR4 and CD36 with CD14 [52] (another cell surface LPS receptor which facilitates interactions between LPS and TLR4). Receptor aggregation/colocalization between CD36 and TLR4 was also detected in retinal pigment epithelial cells [53, 54], the cells that lie between the retina and the choroids (the vascular bed which oxygenates the retina), and are adjacent to the retinal photoreceptors. These examples of CD36–TLR4 interactions provide one possible mechanism whereby CD36 may regulate the NF-κB pathway.
The data above can be summarized as follows: (i) CD36 physically interacts with Src-related PTKs, (ii) CD36 expression modulates the magnitude of oxLDL-mediated NF-κB activation, and (iii) ligand engagement of TLR4 (which mediates NF-κB activation by LPS) induces aggregation with CD36. In addition, we have observed that CD36−/− mice display a muted inflammatory response following LPS injection (V.A. Drover and N.A. Abumrad, unpublished results). Thus, we propose a model (Fig. 5.1) whereby CD36 and its associated PTKs aggregate with TLR4 upon ligand binding. The PTKs may modulate TLR4 signaling to NF-κB via phosphorylation of downstream effector molecules or even the regulatory kinases directly as has been demonstrated previously [55, 56]. The report that CD36 is a signaling bridge for microbial diacylglyceride-stimulated TLR2 [57] supports this model. Further studies in lesions and macrophages from WT and CD36−/− mice are required to test the validity of this model and the potential role of CD36 in mediating atherosclerosis and inflammation.
Scavenger Receptor BI
Like CD36, SR-BI is a class B receptor and a type III transmembrane protein. SR-BI appears to have a hairpin topology, is heavily glycosylated, and binds numerous ligands (reviewed in [58]). SR-BI has a broad expression pattern including intestine, macrophages, endothelial cells, and SMCs but is most highly expressed in the liver and steroidogenic tissues where it plays a major role in cholesterol catabolism and hormone synthesis, respectively.
SR-BI is best known as the most physiologically relevant receptor for highdensity lipoprotein (HDL) particles [59]. SR-BI-mediated cholesterol uptake
80 Daisy Sahoo and Victor A. Drover
from HDL occurs by a process called selective uptake; the cholesteryl ester in the core of the HDL particle is selectively extracted and delivered to the cell for subsequent hydrolysis and/or storage. Mice lacking SR-BI have elevated plasma cholesterol, increased HDL particle size, and reduced cholesterol disposal in the liver [60–62]. In vitro, SR-BI also facilitates cholesterol efflux to lipid acceptors such as small unilamellar vesicles and small HDL particles [17, 63]. Together, the biochemical data would predict that SR-BI plays an atheroprotective role by promoting cholesterol efflux into the plasma as HDL and then facilitating hepatic HDL cholesterol disposal. Indeed, numerous studies in atherogenic strains of mice show that SR-BI deficiency promotes atherosclerosis [64–66] while hepatic overexpression is protective [67] (reviewed in [68]; see also Chapter 4).
While structure–function relationships in SR-BI have been extensively characterized, the role of SR-BI in cell signaling and inflammation remains poorly described. However, a few reports use various techniques to directly implicate SR-BI in cell signaling and inflammatory pathways, which may be relevant to atherosclerosis. For instance, Vishnyakova et al. [69] demonstrated that SR-BI is also a receptor for LPS in murine RAW and HeLa cells. Like SR-A, SR-BI may be involved in the clearance of LPS and attenuating the inflammatory response, thus protecting against atherosclerosis.
Serum amyloid A (SAA), a major acute-phase reactant typically transported on HDL, appears to be a ligand for SR-BI. SAA engagement of SRBI in transfected HeLa cells results in enhanced secretion of IL-8 [70], a proinflammatory and atherogenic chemokine (reviewed in [71]). This SR- BI-dependent, SAA-induced increase in IL-8 secretion requires the activation of ERK1/2 and p38 MAPK [70] and suggests a role for SR-BI in these signaling pathways. The observation that ERK1/2 activation by HDL is reduced by SR-BI neutralizing antibodies [72] further support this notion. One scenario whereby this pathway may be physiologically relevant is the ability of dietary cholesterol to raise serum SAA levels [73], which in turn can increase inflammation/IL-8 secretion and thus contribute to lesion formation. However, it is difficult to reconcile the concept that SR-BI mediates an inflammatory role in SAA/MAPK signaling with the observations from genetically modified mice illustrating an atheroprotective role for SR-BI in vivo (discussed earlier). One possibility is that SR-BI is atherogenic in the early stages of lesion development where inflammation may play a more prominent role in disease pathology. This is in agreement with a recent bone marrow transplantation study showing that SR-BI increased lesion development in LDL-R-deficient mice fed a Western diet for only 4 weeks [74].
The best evidence for SR-BI-dependent cell signaling in atherogenesis comes from the nitric oxide (NO) field. NO is critical for arterial tonicity and is an important mediator of atherosclerosis since chronic inhibition of NO production enhances lesion development in hypercholesterolemic rabbits [75]. NO is produced by endothelial NO synthase (eNOS) and several groups have reported that HDL stimulates eNOS activity in both epithelial and CHO cells
Chapter 5. The Role of SRs in Signaling, Inflammation, and Atherosclerosis |
81 |
in an SR-BI-dependent fashion [76, 77]. As a physiologic measure of NO production, Yuhanna et al. [77] measured arterial relaxation in aortic rings ex vivo and found that HDL-mediated relaxation was highly attenuated in aortic rings from SR-BI−/− mice. Although the mechanism of action is unclear, intracellular ceramide levels covary with eNOS activation [76], suggesting that SR-BI may be mediating eNOS activity by regulating levels of this sphingolipid.
Cross talk between SR-BI-mediated eNOS activation and apoptotic cell death adds another layer of complexity to the role of SR-BI in cellular signaling mechanisms. Li et al. [78] recently reported that in the absence of ligand, SR-BI expression activates caspase-8-mediated apoptosis in CHO cells, mouse embryonic fibroblasts, and endothelial cells. Cotransfection with eNOS or incubation with HDL reversed this SR-BI-dependent cell death. The authors suggest that in healthy cells, HDL-SR-BI ligation inhibits inappropriate apoptosis via NO production. Cells in stressful, inflammatory environments have reduced eNOS activity and apoptosis occurs in an attempt to reduce further inflammation and damage to neighboring cells. Elevated mRNA levels of inflammatory markers in the artery in SR-BI−/− mice [79] are consistent with uncontrolled inflammation and support the authors’ hypothesis.
The Lectin-Like oxLDL Receptor-1 (LOX-1)
Also known as OLR1, this SR displays a type II membrane topology and is expressed on endothelial cells, macrophages, SMCs, and platelets. In addition to mLDL, bacteria, and apoptotic cells, LOX-1 can also bind advanced glycation end products and fibronectin, thus suggesting a role in cell adhesion (discussed later). Ligand engagement to LOX-1 appears to be linked to a host of intracellular signaling processes (discussed later) that may affect atherosclerotic progression but prospective studies in genetically modified mice have not been reported. However, the observations that (i) humans and rabbits exhibit LOX-1 accumulation in atherosclerotic lesions [17, 80] and (ii) LOX- 1 mutations in humans are associated with increased cardiovascular disease [55] suggest that LOX-1 is an important mediator of atherosclerosis. As mice lacking LOX-1 are not currently available, most of the definitive studies on the role of LOX-1 in inflammation/signaling utilize a mixed approach of heterologous gene expression, antisense knockdown technology, and specific α- LOX-1 antibodies which block oxLDL binding to the receptor.
One mechanism whereby LOX-1 may mediate atherosclerotic lesion development is through its ability to stimulate leukocyte adhesion following exposure to inflammatory stimuli. In human coronary artery endothelial cells (HCAEC), oxLDL-induced LOX-1 expression caused elevated monocyte chemoattractant protein-1 expression and monocyte adhesion. These effects were attenuated by transfection of the cells with antisense LOX-1, which abrogated LOX-1 expression [81]. Similarly, CHO-K1 cells transfected with a plasmid encoding LOX-1 exhibited increased leukocyte and THP-1 cell adhesion which could be blocked by oxLDL or an α-LOX-1 antibody [82]. In
82 Daisy Sahoo and Victor A. Drover
rats, leukocytes accumulated at sites of inflammation/damage in a LOX-1- dependent fashion with concomitant increases in adhesion molecule expression [83–85]. Although the mechanism(s) responsible for these LOX-1-mediated effects are not known, both MAPK (p38 and p42/p44) and PKC-β pathways appear to be involved in modulating changes in the expression of adhesion molecules [81, 84, 86].
The ability of oxLDL to stimulate other signaling pathways may also be involved in the activation of endothelial cells. Most notable is the ability of oxLDL to elevate levels of reactive oxygen species (ROS). Cominacini et al. [87] documented an oxLDL and LOX-1-dependent increase in intracellular ROS in bovine arterial endothelial cells (BAEC). Levels of NF-κB in the nucleus were also elevated by oxLDL exposure as determined by a qualitative mobility shift assay. Although a causal relationship between ROS production and nuclear NF-κB translocation was not shown in this study, the concept that ROS are upstream signals for NF-κB activation is well documented in the literature [88–92]. The additional observation that ROS levels following oxLDL stimulation rise well before NF-κB activation is detected [87] forms a strong circumstantial case for a LOX-1/ROS-mediated link between oxLDL and the NF-κB pathway in endothelial cells and other cell types such as articular chondrocytes [93].
The LOX-1-dependent rise of ROS in oxLDL-treated BAECs also results in a concomitant reduction of intracellular NO [94]. The authors speculate that oxLDL ligation of LOX-1 in atherosclerotic lesions may explain the impaired endothelium-dependent vasodilation commonly observed in this type of vascular disease. Since ROS are thought to increase oxLDL production in vivo, we see the potential for a vicious loop of LDL oxidation, ROS formation, NO inactivation, and NF-κB-dependent inflammation—all dependent on endothelial LOX-1. Perhaps not surprisingly, the situation may be further exacerbated by the feed-forward upregulation of LOX-1 expression by oxLDL [95, 96].
In addition to ROS signaling, LOX-1 appears to play some role in a myriad of signaling pathways that may be relevant to atherosclerosis. In addition to the role of p38 MAPK in adhesion molecule expression (discussed earlier), oxLDL induces apoptosis in neonatal rat cardiac myocytes in a LOX-1- and p38 MAPK-dependent fashion [97]. In cultured rat chondrocytes and HCAECs, oxLDL induces cell death via PI3 kinase/Akt [98] and NF-κB pathways [99], respectively; in each instance, cell death could be blocked by α-LOX-1 antibodies or abrogating LOX-1 expression. Given the complexity of the signaling pathways stimulated by oxLDL via LOX-1, significant cross talk appears likely.
Since LOX-1 is thought to be a major SR on endothelial cells, much of the work described herein has used primary or cultured endothelial cells. However, it is important to note that many of the pathways implicated in LOX-1 signaling are present in macrophages. LOX-1 is expressed on macrophages and oxLDL ligation may affect inflammation/apoptosis in this cell type in addition to endothelial cell activation. It is also likely that some of these signaling pathways terminate at the genes encoding adhesion molecules and other cell typespecific genes in the inflammatory/atherosclerotic expression profile (Fig. 5.2).
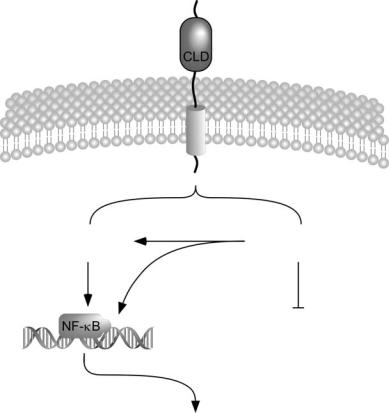
Chapter 5. The Role of SRs in Signaling, Inflammation, and Atherosclerosis |
83 |
LOX-1
MAPK, |
Reactive |
PKC, P13-K |
oxygen species |
Nitric oxide
LOX-1, TNF-α, CD40/CD40L (inflammation)
MCP-1, ICAM, VCAM (adhesion)
MMP-1, MMP-3, MMP-9 (plaque rupture)
FIGURE 5.2. Intracellular signaling pathways following LOX-1 engagement by oxLDL in endothelial cells. LOX-1 is a type II integral membrane protein and contains a C-type lectin-like domain (CLD). Oxidized LDL engagement of LOX-1 causes the increased expression of numerous genes whose protein products are involved in inflammation, monocyte adhesion, and plaque rupture. These changes in gene expression occur as a result of the activation of numerous intracellular signaling pathways including mitogen-activated protein kinase (MAPK), protein kinase C (PKC), and PI3 kinase (PI3-K), as well as through the production of reactive oxygen species (ROS). It is likely that many of these signaling pathways act directly or indirectly via the NF-κB pathway. In addition, increased ROS production inactivates intracellular nitric oxide (NO), which may result in impaired vasodilation commonly observed in patients with atherosclerosis.
A major hurdle in the elucidation of these pathways is the lack of mice with a disrupted LOX-1 gene. Tissue/cell type-specific gene ablation may be necessary to disentangle the plethora of intracellular signals initiated by LOX-1 ligand engagement.
84 Daisy Sahoo and Victor A. Drover
Concluding Remarks
As the field of SR biology expands, our understanding of the functions of these proteins moves beyond the clearance and utilization of a particular ligand. As should now be evident, SRs are as promiscuous in their use of intracellular signaling pathways as they are in their cell surface ligands. Our understanding of these pathways and their role in the pathology of atherosclerosis is still a developing area of research, especially as many SRs have only recently been classified in this protein family. In addition, determining the net effect of engaging multiple SRs with a common ligand such as mLDL/oxLDL will be a challenging task. This will be further complicated by the fact that multiple SRs can affect the same signaling pathways. For instance, LOX-1 shares a functional link to NF-κB and NO with CD36 and SR-BI, respectively. Similarly, CD36 and SR-A both interact with Src-family PTKs.
While examining the effects of multilocus gene disruption may provide an overall measure of the relative contribution of the SRs to atherogenesis, determining the relevant mechanisms will require careful experimental design and attention to the effective cell type. In addition to the relative distribution of the SRs in critical cell types such as endothelial cells and monocytes/macrophages, the ability of a ligand or nutritional regimen to regulate SR levels and concomitant signaling pathways must be considered. The continued use of genetically modified mice and tissue-specific gene ablation are thus likely to play important roles in answering the remaining questions in this field.
References
1.Skalen K, Gustafsson M, Rydberg EK, Hulten LM, Wiklund O, Innerarity TL, Boren J: Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature 417: 750–754, 2002.
2.Stocker R, Keaney JF Jr: Role of oxidative modifications in atherosclerosis. Physiol Rev 84: 1381–1478, 2004.
3.Stokes KY, Cooper D, Tailor A, Granger DN: Hypercholesterolemia promotes inflammation and microvascular dysfunction: role of nitric oxide and superoxide. Free Radic Biol Med 33: 1026–1036, 2002.
4.Kannel WB: Status of risk factors and their consideration in antihypertensive therapy. Am J Cardiol 59: 80A–90A, 1987.
5.Abumrad N, Coburn C, Ibrahimi A: Membrane proteins implicated in longchain fatty acid uptake by mammalian cells: CD36, FATP and FABPm. Biochim Biophys Acta 1441: 4–13, 1999.
6.Leitinger N: Oxidized phospholipids as modulators of inflammation in atherosclerosis. Curr Opin Lipidol 14: 421–430, 2003.
7.Eriksson EE, Xie X, Werr J, Thoren P, Lindbom L: Importance of primary capture and L-selectin-dependent secondary capture in leukocyte accumulation in inflammation and atherosclerosis in vivo. J Exp Med 194: 205–218, 2001.
8.Janeway CA Jr, Medzhitov R: Innate immune recognition. Annu Rev Immunol 20: 197–216, 2002.
Chapter 5. The Role of SRs in Signaling, Inflammation, and Atherosclerosis |
85 |
9.Peiser L, Mukhopadhyay S, Gordon S: Scavenger receptors in innate immunity. Curr Opin Immunol 14: 123–128, 2002.
10.Goldstein JL, Brown MS: The low-density lipoprotein pathway and its relation to atherosclerosis. Annu Rev Biochem 46: 897–930, 1977.
11.Nagy L, Tontonoz P, Alvarez JG, Chen H, Evans RM: Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell 93: 229–240, 1998.
12.Frostegard J, Ulfgren AK, Nyberg P, Hedin U, Swedenborg J, Andersson U, Hansson GK: Cytokine expression in advanced human atherosclerotic plaques: dominance of pro-inflammatory (Th1) and macrophage-stimulating cytokines. Atherosclerosis 145: 33–43, 1999.
13.Uyemura K, Demer LL, Castle SC, Jullien D, Berliner JA, Gately MK, Warrier RR, Pham N, Fogelman AM, Modlin RL: Cross-regulatory roles of inter- leukin(IL)-12 and IL-10 in atherosclerosis. J Clin Invest 97: 2130–2138, 1996.
14.Falk E, Shah PK, Fuster V: Coronary plaque disruption. Circulation 92: 657–671, 1995.
15.Brown AM, Baker PW, Gibbons GF: Changes in fatty acid metabolism in rat hepatocytes in response to dietary n-3 fatty acids are associated with changes in the intracellular metabolism and secretion of apolipoprotein B-48. J Lipid Res
38:469–481, 1997.
16.Goldstein JL, Ho YK, Basu SK, Brown MS: Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc Natl Acad Sci USA 76: 333–337, 1979.
17.Suzuki H, Kurihara Y, Takeya M, Kamada N, Kataoka M, Jishage K, Ueda O, Sakaguchi H, Higashi T, Suzuki T, Takashima Y, Kawabe Y, Cynshi O, Wada Y, Honda M, Kurihara H, Aburatani H, Doi T, Matsumoto A, Azuma S, Noda T, Toyoda Y, Itakura H, Yazaki Y, Horiuchi S, Takahashi K, Kruijt JK, van Berkel TJ, Steinbrecher UP, Ishibashi S, Maeda N, Gordon S, Kodama T et al: A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature 386: 292–296, 1997.
18.Rohrer L, Freeman M, Kodama T, Penman M, Krieger M: Coiled-coil fibrous domains mediate ligand binding by macrophage scavenger receptor type II. Nature 343: 570–572, 1990.
19.Murphy JE, Tedbury PR, Homer-Vanniasinkam S, Walker JH, Ponnambalam S: Biochemistry and cell biology of mammalian scavenger receptors. Atherosclerosis
182:1–15, 2005.
20.Kunjathoor VV, Febbraio M, Podrez EA, Moore KJ, Andersson L, Koehn S, Rhee JS, Silverstein R, Hoff HF, Freeman MW: Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J Biol Chem 277: 49982–49988, 2002.
21.Haworth R, Platt N, Keshav S, Hughes D, Darley E, Suzuki H, Kurihara Y, Kodama T, Gordon S: The macrophage scavenger receptor type A is expressed by activated macrophages and protects the host against lethal endotoxic shock. J Exp Med 186: 1431–1439, 1997.
22.Moore KJ, Kunjathoor VV, Koehn SL, Manning JJ, Tseng AA, Silver JM, McKee M, Freeman MW: Loss of receptor-mediated lipid uptake via scavenger receptor A or CD36 pathways does not ameliorate atherosclerosis in hyperlipidemic mice. J Clin Invest 115: 2192–2201, 2005.
86Daisy Sahoo and Victor A. Drover
23.Babaev VR, Gleaves LA, Carter KJ, Suzuki H, Kodama T, Fazio S, Linton MF: Reduced atherosclerotic lesions in mice deficient for total or macrophage-specific expression of scavenger receptor-A. Arterioscler Thromb Vasc Biol 20: 2593–2599, 2000.
24.Herijgers N, de Winther MP, Van Eck M, Havekes LM, Hofker MH, Hoogerbrugge PM, Van Berkel TJ: Effect of human scavenger receptor class A overexpression in bone marrow-derived cells on lipoprotein metabolism and atherosclerosis in low density lipoprotein receptor knockout mice. J Lipid Res 41: 1402–1409, 2000.
25.Whitman SC, Rateri DL, Szilvassy SJ, Cornicelli JA, Daugherty A: Macrophagespecific expression of class A scavenger receptors in LDL receptor(−/−) mice decreases atherosclerosis and changes spleen morphology. J Lipid Res 43: 1201–1208, 2002.
26.Ashkenas J, Penman M, Vasile E, Acton S, Freeman M, Krieger M: Structures and high and low affinity ligand binding properties of murine type I and type II macrophage scavenger receptors. J Lipid Res 34: 983–1000, 1993.
27.Fong LG, Le D: The processing of ligands by the class A scavenger receptor is dependent on signal information located in the cytoplasmic domain. J Biol Chem 274: 36808–36816, 1999.
28.Ricci R, Sumara G, Sumara I, Rozenberg I, Kurrer M, Akhmedov A, Hersberger M, Eriksson U, Eberli FR, Becher B, Boren J, Chen M, Cybulsky MI, Moore KJ, Freeman MW, Wagner EF, Matter CM, Luscher TF: Requirement of JNK2 for scavenger receptor A-mediated foam cell formation in atherogenesis. Science 306: 1558–1561, 2004.
29.Pollaud-Cherion C, Vandaele J, Quartulli F, Seguelas MH, Decerprit J, Pipy B: Involvement of calcium and arachidonate metabolism in acetylated-low-density- lipoprotein-stimulated tumor-necrosis-factor-alpha production by rat peritoneal macrophages. Eur J Biochem 253: 345–353, 1998.
30.Whitman SC, Daugherty A, Post SR: Regulation of acetylated low density lipoprotein uptake in macrophages by pertussis toxin-sensitive G proteins. J Lipid Res 41: 807–813, 2000.
31.Kienast J, Padro T, Steins M, Li CX, Schmid KW, Hammel D, Scheld HH, van de Loo JC: Relation of urokinase-type plasminogen activator expression to presence and severity of atherosclerotic lesions in human coronary arteries. Thromb Haemost 79: 579–586, 1998.
32.Hsu HY, Hajjar DP, Khan KM, Falcone DJ: Ligand binding to macrophage scavenger receptor-A induces urokinase-type plasminogen activator expression by a protein kinase-dependent signaling pathway. J Biol Chem 273: 1240–1246, 1998.
33.Miki S, Tsukada S, Nakamura Y, Aimoto S, Hojo H, Sato B, Yamamoto M, Miki Y: Functional and possible physical association of scavenger receptor with cytoplasmic tyrosine kinase Lyn in monocytic THP-1-derived macrophages. FEBS Lett 399: 241–244, 1996.
34.Miki S, Horikawa K, Nishizumi H, Suemura M, Sato B, Yamamoto M, Takatsu K, Yamamoto T, Miki Y: Reduction of atherosclerosis despite hypercholesterolemia in Lyn-deficient mice fed a high-fat diet. Genes Cells 6: 37–42, 2001.
35.Amersfoort ESV, Van Berkel TJC, Kuiper J: Clearance of lipopolysaccharide in scavenger receptor knockout mice. J Leukocyte Biol 210 (Suppl): 48, 1996.
36.Yanai H, Chiba H, Morimoto M, Abe K, Fujiwara H, Fuda H, Hui SP, Takahashi Y, Akita H, Jamieson GA, Kobayashi K, Matsuno K: Human CD36
Chapter 5. The Role of SRs in Signaling, Inflammation, and Atherosclerosis |
87 |
deficiency is associated with elevation in low-density lipoprotein-cholesterol. Am J of Med Gen 93: 299–304, 2000.
37.Febbraio M, Hajjar DP, Silverstein RL: CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest 108: 785–791, 2001.
38.Febbraio M, Podrez EA, Smith JD, Hajjar DP, Hazen SL, Hoff HF, Sharma K, Silverstein RL: Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest 105: 1049–1056, 2000.
39.Febbraio M, Abumrad NA, Hajjar DP, Sharma K, Cheng W, Pearce SF, Silverstein RL: A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J Biol Chem 274: 19055–19062, 1999.
40.Watt MJ, Southgate RJ, Holmes AG, Febbraio MA: Suppression of plasma free fatty acids upregulates peroxisome proliferator-activated receptor (PPAR) alpha and delta and PPAR coactivator 1 alpha in human skeletal muscle, but not lipid regulatory genes. J Mol Endocrinol 33: 533–544, 2004.
41.Malaud E, Hourton D, Giroux LM, Ninio E, Buckland R, McGregor JL: The terminal six amino-acids of the carboxy cytoplasmic tail of CD36 contain a functional domain implicated in the binding and capture of oxidized low-density lipoprotein. Biochem J 364: 507–515, 2002.
42.Puente Navazo MD, Daviet L, Ninio E, McGregor JL: Identification on human CD36 of a domain (155–183) implicated in binding oxidized low-density lipoproteins (ox-LDL). Arterioscler Thromb Vasc Biol 16: 1033–1039, 1996.
43.Robbesyn F, Salvayre R, Negre-Salvayre A: Dual role of oxidized LDL on the NF-kappaB signaling pathway. Free Radic Res 38: 541–551, 2004.
44.Lipsky RH, Eckert DM, Tang Y, Ockenhouse CF: The carboxyl-terminal cytoplasmic domain of CD36 is required for oxidized low-density lipoprotein modulation of NF-kappaB activity by tumor necrosis factor-alpha. Recept Signal Transduct 7: 1–11, 1997.
45.Janabi M, Yamashita S, Hirano K, Sakai N, Hiraoka H, Matsumoto K, Zhang Z, Nozaki S, Matsuzawa Y: Oxidized LDL-induced NF-kappa B activation and subsequent expression of proinflammatory genes are defective in monocytederived macrophages from CD36-deficient patients. Arterioscler Thromb Vasc Biol 20: 1953–1960, 2000.
46.Bull HA, Brickell PM, Dowd PM: Src-related protein tyrosine kinases are physically associated with the surface antigen CD36 in human dermal microvascular endothelial cells. FEBS Lett 351: 41–44, 1994.
47.Moore KJ, El Khoury J, Medeiros LA, Terada K, Geula C, Luster AD, Freeman MW: A CD36-initiated signaling cascade mediates inflammatory effects of betaamyloid. J Biol Chem 277: 47373–47379, 2002.
48.Lee H, Woodman SE, Engelman JA, Volonte D, Galbiati F, Kaufman HL, Lublin DM, Lisanti MP: Palmitoylation of caveolin-1 at a single site (Cys-156) controls its coupling to the c-Src tyrosine kinase: targeting of dually acylated molecules (GPI-linked, transmembrane, or cytoplasmic) to caveolae effectively uncouples c-Src and caveolin-1 (TYR-14). J Biol Chem 276: 35150–35158, 2001.
49.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S: Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol 162: 3749–3752, 1999.