

биохимия атеросклероза
.pdf210 Rita Kohen Avramoglu et al.
a close relative of the ob/ob mouse, displays a similar phenotype although the genetic defect responsible appears to be a mutation in the leptin receptor gene, rather than in leptin itself. While the ob/ob mouse displays hyperplasia of pancreatic cells, the db/db mouse displays evidence of β-cell degranulation and apoptosis. Both the ob/ob and db/db mice display abnormal lipid profiles with hypertriglyceridemia, although in the ob/ob mouse, there also appears to be a defect in the catabolism of apoAI and apoAII leading to increased HDL levels [45]. It has recently been found that glucose-6-phosphate levels are elevated in both ob/ob and db/db mice [46]. This may partly account for the dysregulation of glucose metabolism and increased hepatic VLDL production in these animals. While treatment with thiazolidinediones improves the insulin sensitivity of muscle tissue and the fatty liver phenotype in ob/ob mice, hepatic VLDL production appears less sensitive to insulin treatment [47–49].
One of the earliest rat models used, the Zucker fatty rat (fa/fa), also carries a mutation in the leptin receptor gene [50]. This model exhibits hyperglycemia, hyperinsulinemia, type 2 diabetes, hypertriglyceridemia, hypercholesterolemia, in addition to increased appetite [51]. Early studies showed neointimal hyperplasia following injury in the diabetic animals compared with lean controls [52]. One of the striking features of Zucker rats is the accumulation of TG-rich lipoproteins such as chylomicrons in the plasma [53]. It has been shown that there is an elevation in MTP mRNA in Zucker rats compared with lean controls that may partly contribute to the VLDL overproduction [54]. Following insulin treatment, Zucker rats show resistance to completely normalize lipoprotein profiles [55]. Recently, treatment with thiazolidinediones showed an improvement in insulin-induced FFA clearance, improved FFA usage during fasting, improved glucose homeostasis, a decrease in hepatic TG production, and increased TG clearance [56, 57]. However, statin treatment resulted in marked decreases in plasma TG, total cholesterol, and total HDL. The currently available data suggests that in this model plasma TG but not cholesterol may be dependent on plasma insulin levels [58].
The homozygous JCR:LA corpulent rat (cp/cp) exhibits obesity, hypertriglyceridemia, hyperinsulinemia, and impaired glucose tolerance [59–61]. In these animals, insulin resistance appears to be more severe in homozygous males, while females and lean animals do not appear to develop atherosclerosis spontaneously. There is also enhanced secretion of VLDL particles that results from the conversion of dietary carbohydrate into TG [62].
Corroborating evidence from other animal models include the Watanabe heritable hyperlipidemic (WHHL) rabbit model of familial hypercholesterolemia. This animal model exhibits hyperinsulinemia and insulin resistance, and the addition of a small amount of cholesterol to the diet results in elevated plasma lipids and severe cardiovascular disease [63]. Administration of thiazolidinediones in combination with HMG-CoA reductase inhibitors lowers plasma lipid levels and ameliorates hypertension and insulin sensitivity [64–67]. Transgenic WHHL rabbits overexpressing human lipoprotein lipase (LPL) also
Chapter 11. Lipoprotein Metabolism in Insulin-Resistant States |
211 |
showed improvements in hypertriglyceridemia, hypercholesterolemia, and obesity suggesting potential therapeutic benefits of LPL [68].
The transgenic mouse model using overexpression of the A1 adenosine receptor in adipose tissue has implicated this receptor in the metabolism of intracellular fat accumulation, FFA metabolism, and plasma glucose regulation [69–72]. Interestingly, although both control and transgenic animals were of the same size and body composition, the transgenic animals exhibited lower plasma FFA, and failed to develop glucose intolerance in oral glucose tolerance tests. Another genetically altered model, the ApoB/BATless mouse, is a cross between a human apoB transgenic mouse and a brown adipose tissue (BAT) knockout (KO) mouse. This model exhibits peripheral insulin resistance, resulting in obesity, hypertriglyceridemia, hypercholesterolemia, and hyperinsulinemia once placed on a high-fat diet [73]. Although increases in apoB levels were seen in the apoB/BATless mice, the mRNA levels of both MTP, as well as apoB, were similar between these and control mice expressing only the human apoB transgene. This suggests that VLDL assembly and secretion were instead regulated posttranslationally.
Several rodent models of diet-induced insulin resistance have also been developed and employed to enhance our understanding of the underlying mechanisms associated with complications due to dyslipidemia. The “sand rat” (Psammomys obesus), a gerbil native to the desert regions of the eastern Mediterranean and northern Africa, spontaneously develops obesity and insulin resistance when fed standard rodent chow instead of its customary diet of succulent plants [39, 41]. These animals exhibit signs of hyperglycemia, insulin resistance, dyslipidemia, obesity, and hyperphagia [74–77]. The defect is now believed to at least partly result from apoptosis of pancreatic β-cells. There is also evidence of insulin receptor (IR) signaling pathway dysfunction [44, 78], hepatic insulin resistance [79], and insensitivity to the satiety factor leptin [80]. Characterization of the lipid distribution within these animals showed significant increases in circulating VLDL and LDL in both hyperinsulinemic, as well as hyperinsulinemic and hyperglycemic animals [81]. Elevated levels of protein kinase C (PKC) epsilon in skeletal muscle are thought to contribute to the development of insulin resistance in these animals [82]. In addition, the novel protein factor, beacon, was also found to be differentially expressed in the brain of obese and control animals [83].
A fructose-fed Syrian golden hamster model has been developed in our laboratory and is increasingly used as a simple dietary model of insulin resistance and metabolic dyslipidemia. This diet-induced model of insulin resistance has been extensively characterized [84], and is of interest as its lipoprotein metabolism more closely resembles that of humans compared to other rodent models [85–88]. Hamsters develop hyperlipidemia and atherosclerosis in response to a modest increase in dietary cholesterol and saturated fat and can be made obese, hypertriglyceridemic, and insulin-resistant by fructose feeding [84, 89, 90]. Fructose-induced metabolic dyslipidemia is also usually accompanied by whole-body insulin resistance and reduced hepatic
212 Rita Kohen Avramoglu et al.
insulin sensitivity [84, 91]. Fructose feeding for a 2-week period induced significant increases in plasma TG, cholesterol, and FFA. Induction of insulin resistance was accompanied by a considerable rise in the in vivo production of hepatic VLDL-apoB and –TG [84]. These data suggest overall improved efficiency of VLDL assembly in animals fed a high-fructose diet. This may be due to the increased intracellular stability and availability of apoB, elevated levels of available neutral lipid, or increased MTP mass or activity [92, 93]. A more detailed discussion of mechanisms of metabolic dyslipidemia in the fructose-fed hamster model will appear later in this chapter.
Molecular Mechanisms of Insulin Resistance and
Metabolic Dyslipidemia: Perturbations in Insulin
Signaling Cascades
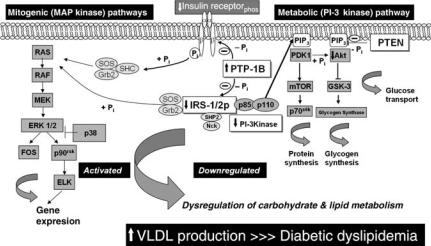
Under normal metabolic conditions, the binding of insulin to its plasma membrane receptor initiates a cascade of events, beginning with receptor autophosphorylation and activation of receptor tyrosine kinases (see Fig. 11.1) [94, 95]. Receptor autophosphorylation, in turn, results in tyrosine
FIGURE 11.1. Postulated link between hepatic insulin signaling and downstream VLDL-apoB secretion. Insulin induces signal transduction via two major signaling pathways: the mitogenic, MAP-kinase pathway and the metabolic, PI 3-kinase pathway. Under normal conditions, insulin acutely reduces apoB secretion. Under conditions of insulin resistance, however, there is reduced sensitivity to inhibitory action of insulin on apoB resulting in enhanced VLDL secretion. Enhanced expression of PTP-1B, a key negative regulator of insulin signaling, may be a key initiating factor in inducing hepatic insulin resistance and consequently increased VLDL synthesis and secretion.
Chapter 11. Lipoprotein Metabolism in Insulin-Resistant States |
213 |
phosphorylation of adaptor proteins such as members of the insulin receptor substrate (IRS) family (IRS-1/2/3/4), and Shc [96–100]. Insulin signaling is regulated by the activity of phosphotyrosyl-protein phosphatases (PTPases) through dephosphorylation of the insulin receptor, IRS-1, IRS-2, and Shc leading to modulation of insulin action downstream of the receptor [101]. IRS-1 and IRS-2 are adaptor proteins for src homology 2 (SH2)- domain containing signaling proteins including the Grb-2–SOS complex, SHP2, Nck, and PI-3 kinase [102–105]. The production of 3′-phospholipids such as PI-3,4,5-P3 (PIP3) is dependent on activation of PI3-kinase. Binding of IRS to p85, the regulatory subunit of PI-3 kinase, activates the PI-3 kinase–PKB/Akt pathway, which is necessary for insulin action on glucose transport and glycogen synthesis [97, 106, 107].
Insulin resistance is thought to develop as a result of defects downstream of the IR activation. Because of the complexity of the numerous branches of the insulin signaling cascade, cross talk between signaling molecules, and the effects of detrimental environmental factors, attempts at understanding the precise mechanisms involved in the development of insulin-resistant states has proven challenging. Defects in insulin signaling appear to occur primarily in insulin sensitive tissues, namely liver, adipose, and muscle. Among obese and type 2 diabetic individuals, significant decreases in IRS-1-associated tyrosine phosphorylation and PI3-kinase activity have been observed in skeletal muscle and adipocytes; the two tissues predominantly targeted by insulin [107–111]. Obese and hyperinsulinemic individuals exhibited decreased IR expression level and activity, in addition to decreased tyrosine kinase activity, in both skeletal muscle and adipocytes [112, 113], while patients with type 2 diabetes exhibit reduced insulin signaling in skeletal muscle and the liver [112, 114]. In lean type 2 diabetic subjects, insulin-stimulated PI-3 kinase activity was also decreased suggesting that a defect in insulin signaling could contribute to insulin resistance [111]. Similar findings have been reported in numerous genetic [115–119] and induced [120, 121] rodent models of obesity. Targeted disruption of the IR and IRS-1 or IRS-2 [122–127] have all shown that an insensitivity to the effects of insulin in a combination of tissues including muscle, liver, and adipose tissue can lead to insulin resistance and type 2 diabetes [107].
Defects in molecules of the insulin-signaling pathway are now thought to be a major mechanism involved in the development of insulin resistance. Perturbations are documented in both arms of the insulin-signaling pathway, the PI-3 kinase and the mitogen-activated protein (MAP) kinase cascades. Activated PI3-kinase generates 3′-phospholipids, including PIP3, which mediates many cellular responses of insulin [128, 129]. PIP3 levels are attenuated by the action of a lipid phosphatase, PTEN [107]. Perturbations in PTEN can thus have important consequences on the PI-3 kinase signaling cascade.
Many gene regulatory events induced by receptor-associated kinases (such as IR) are mediated by the MAP kinase cascade (reviewed in Refs.
214 Rita Kohen Avramoglu et al.
[130–132]). Several families of the MAP kinases exist including specifically extracellular signal-regulated kinase (ERK) 1/2, c-jun kinase (JNK), and p38. There is increasing evidence that members of the MAP kinase family of serine–threonine kinases may contribute to the development of insulin resistance. IRS-1 contains many potential MAP kinase phosphorylation sites [133], and it has been observed that certain MAP kinases are capable of phosphorylating and inactivating IRS-1 [134–136]. Furthermore, insulin stimulation is capable of activating members of the MAP kinase family, specifically ERK1/2, JNK, and p38 [137–139]. In human skeletal muscle, insulin signaling to ERK1/2 was normal from patients with type 2 diabetes compared with healthy control subjects, despite a diminished insulin responsiveness of components required for the metabolic effects of insulin such as components of the PI-3 kinase and AKT pathways [140, 141]. In other models an increased basal level of phosphorylation of ERK1/2, JNK, or p38 was observed in adipocytes from type 2 diabetic subjects. This was found to associate with a reduction in IRS-1 and glucose transporter 4 (GLUT4), suggesting that elevated MAP kinase activation may contribute to the type 2 diabetic condition [142, 143]. Interestingly, tumor necrosis factor (TNF) α-mediated insulin resistance appears to be mediated by the MAP/ERK kinase (MEK) 1/2-p42/44 MAP kinase pathway [144]. Overall, these very intriguing observations indicate a critical role for activated MAP kinase cascade in insulin resistance.
Recent evidence has also implicated alterations in expression of cellular phosphatases in the development of insulin resistance. Of particular interest is the PTP-1B, a member of the PTPase family of enzymes, which is widely expressed in insulin sensitive tissues [145]. PTP-1B dephosphorylates the IR in vitro, and as well inducing the downregulation of IRS-1 and insulinstimulated PI-3 kinase activity [146–148]. Increased PTP-1B mass and activity have been shown to increase with carbohydrate-induced insulin resistance; while normalization of PTP-1B mass and activity result in the reversal of this type of insulin resistance [149, 150]. PTP-1B KO mice exhibit increased sensitivity toward insulin-induced IR and IRS-1 tyrosine phosphorylation, and are resistant to obesity [151]. Additional evidence of PTP-1B involvement in insulin resistance is seen in primary hepatocytes from an insulin resistant hamster where PTP-1B mass and activity were shown to increase, with a concomitant decrease in PI-3-K and PKB/Akt phosphorylation [93]. Interestingly, a significant increase in PTP-1B protein mass was seen following 2 days of insulin treatment in primary hepatocytes, which was then accompanied by a decrease in IR mass and phosphorylation. PTP-1B has also been shown to influence leptin, an important hormone known to be involved in the regulation of appetite and body mass [152]. In transfection studies, as well as in transgenic animals, PTP-1B has been shown to dephosphorylate the leptin receptor-associated kinase (Janus kinase 2, JAK2) [153–155]. Conversely, in PTP-1B deficient mice, there was an enhanced response toward leptin-mediated loss of body weight.
Chapter 11. Lipoprotein Metabolism in Insulin-Resistant States |
215 |
Adipose Tissue Insulin Resistance, Dysregulation of Fatty Acid Storage and Enhanced Free Fatty Acid Flux
Free fatty acid (FFA), whether obtained through dietary sources or produced via de novo lipogenesis, may generally undergo three fates: intracellular storage by adipose tissue in the form of TG [156]; export into the plasma in the form of VLDL-TG; or use as an energy source via FFA oxidation. Although FFA may also be stored in nonadipocyte cells as TG, this is usually a source of lipotoxicity [157]. Nonadipose TG storage usually occurs in situations where FFA export exceeds the catabolic capacity of metabolic tissues, or storage in adipose tissue is impaired [156]. Over 30% of energy expenditure occurs in skeletal muscle, which modulates the balance between stored lipids and energy utilization [158]. FFA oxidation is induced through both reductions in lipogenic gene expression and upregulation in genes controlling lipid oxidation [157]. These gene alterations are achieved through sophisticated nutrient sensors, and subsequent nuclear signaling cascades.
The net concentration of FFA in the plasma results from a delicate equilibrium between enzyme-regulated lipolysis of plasma triglyceride-rich lipoproteins such as VLDL, and FFA uptake by peripheral tissues. Hormonal regulation of extracellular and intracellular factors affecting FFA metabolism is currently not well understood. Under normal conditions, insulin stimulates postprandial uptake of glucose, as well as FFA esterification and storage, and can play a major role in the suppression of hormone sensitive lipase (HSL), the enzyme that is the major regulator of FFA release from adipose tissue [159]. Insulin and glucose are also believed to stimulate LPL activity, primarily from adipose tissue. LPL activity provides an essential first step in the delivery of FFA to adipose tissue for storage, as well as plasma TG removal.
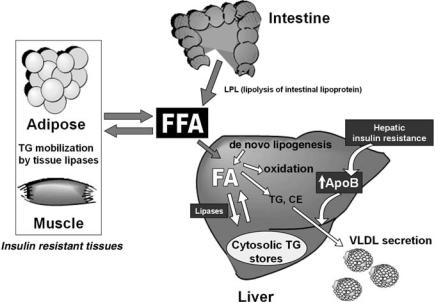
Both fasting and postprandial levels of FFA are often elevated in obese and insulin-resistant individuals. In the insulin-resistant state, there is an increase in release of FFA from adipose tissue concomitant with a decrease in uptake by muscle tissue. This results in an increased influx of FFA into the liver. This “vicious cycle” may result in an attenuation in insulin signaling in these tissues and may exacerbate insulin resistance (see Fig. 11.2) [160, 161]. In muscle, insulin resistance may be mediated by soluble factors; likely derived from fat tissue [162]. Additional factors may influence FFA flux in insulin insensitive conditions. PPAR-gamma activation can lower circulating FFA and increase oxidation through stimulation of adiponectin production. Adiponectin stimulation results in decreased liver TG and increased insulin sensitivity [163]. Although these processes are by far exceeded by postprandial availability of FFA, insulin is known to stimulate enzymes involved in de novo lipogenesis. Generally under conditions where influx of fat to the liver exceeds efflux, there is excess hepatic fatty acid uptake, synthesis and secretion; and hepatic steatosis can ensue alongside insulin resistance [164]. In addition, overloading of white adipose tissue beyond its storage capacity can
216 Rita Kohen Avramoglu et al.
FIGURE 11.2. Mechanisms of diabetic dyslipidemia in insulin-resistant tissues. Insulin resistance results in enhanced TG lipolysis by adipocytes and decreased FFA absorption resulting in an enhanced FFA flux into peripheral tissues. Increased lipid availability resulting from elevated FFA flux combined with hepatic and intestinal insulin resistance appear to lead to a considerable overproduction of both hepaticallyand intestinally derived apoB-containing lipoprotein particles. These events may, in turn, exacerbate fasting and postprandial metabolic dyslipidemia.
also lead to lipid disorders in skeletal and cardiac muscles, and pancreas, illustrating the importance of functional adipocytes [165].
The true underlying culprit to adipose dysfunction and metabolic syndrome may be a chronic positive energy balance—where dysregulation of adipose tissue metabolism may be a catalyst for the chain of events leading to metabolic dyslipidemia [166]. White adipose tissue is now well recognized as an endocrine tissue producing multiple hormones. Plasma levels of these adipocyte hormones, or adipokines, are altered in obese, insulin-resistant, and diabetic subjects [165]. In this way, adipose tissue participates in the regulation of glucose and insulin metabolism through the release of adipokines including leptin, adiponectin, and resistin [167].
The insulin-sensitizing hormone adiponectin is found in lower concentrations corresponding with greater adiposity [166, 168, 169], and is currently considered as an important link between obesity and insulin resistance. Decreased adipocyte differentiation, resulting in decreased adiponectin expression may be involved in the whole-body insulin resistance associated with obesity [170, 171]. Adiponectin is associated with inflammatory markers
Chapter 11. Lipoprotein Metabolism in Insulin-Resistant States |
217 |
through underlying obesity, whereas association of resistin with inflammatory markers appear to be less clear [172]. This connection is further illustrated by significant weight loss with improvements in insulin resistance, atherosclerosis, and cardiovascular disease usually accompanied by increases in adiponectin levels [169, 173]. It has been further proposed that improvements in insulin resistance observed following bariatric surgery are modulated by changes in adipocytokine levels via loss of adipose mass, supporting a role for adiponectin and resistin in the development of insulin resistance and diabetes [162, 167].
Resistin is a protein secreted from adipose tissue that is also thought to provide a link between obesity, insulin sensitivity, and diabetes [174–176]. Although resistin may have an important involvement in insulin resistance, animal and human studies currently show conflicting results [167]. The regulation of resistin expression by agents known to modulate insulin sensitivity does not always support the association between resistin and obe- sity-induced insulin resistance [174]. It has been surprisingly found that high serum resistin levels can predict future increases in percent body fat, but do not predict insulin resistance in humans [177]. On the other hand, decreases in resistin levels may contribute to improving insulin action [175]. Consistent with this observation, high-fat feeding is known to induce the development of obesity and insulin resistance alongside increases in resistin expression. In addition, there are also positive correlations between glycemia and resistin gene expression [174]. Resistin protein level is elevated in obese mice and may be decreased by the use of insulin-sensitizing thiazolidinediones, while immunoneutralization of resistin improves insulin sensitivity in obese mice.
Dysregulation of Fatty Acid Oxidation in Insulin
Resistance
Metabolic dyslipidemia of insulin-resistant states may arise from increased peripheral FFA exposure due to reduced FFA oxidation [178]. Impaired FFA oxidation may contribute to excess TG accumulation, and may mediate numerous adverse effects of metabolic insulin resistance syndrome [179]. The associated inflammation can lead to numerous dysfunctions in lipid and lipoprotein metabolism, alongside marked reductions in lipid oxidation [180]. In the insulin-resistant state, there is a failure of skeletal muscle to shift between glucose and lipid utilization in fasting and insulin-stimulated states. In addition, there is also a failure to suppress FFA efflux from adipose tissue, as well as an impaired FFA efflux and storage balance [181]. Thus TG accumulation in skeletal muscle results from a decrease in FFA oxidation, as well as increased FFA uptake [157].
Mechanisms of the reductions in FFA oxidation include reduced carnitine palmitoyl transferase (CPT) activity and the resultant increase in intracellular
218 Rita Kohen Avramoglu et al.
malonyl-CoA [157], chronic hyperglycemia and hyperinsulinemia [182], decreased expression of PPARgamma coactivator-1alpha (PGC-1alpha) [179], acquired or inherited defects in mitochondrial fatty acid oxidation [183], and decreased expression of mitochondria and mitochondrial enzymes in muscle [179]. Plasma TG levels increase following the suppression of FFA oxidation in patients with type 2 diabetes, where lipid oxidation and insulinmediated glucose oxidation are reduced [178, 180].
Nuclear Signaling Cascades Regulating Fatty Acid
Storage and Oxidation
The physiological regulation of lipogenic gene expression and metabolism is mediated by nuclear hormone receptors, which function as hormone and nutrient-regulated transcription factors that bind DNA [158]. Metabolic nuclear receptors such as receptors for oxysterols (LXRs), bile acids (FXR), and FFA (through PPAR) regulate key aspects of cellular and whole-body sterol homeostasis. These processes include lipoprotein synthesis, lipoprotein uptake by peripheral tissues, cholesterol absorption, bile acid synthesis, and reverse cholesterol transport [184]. The liver nuclear receptors also act as nutrient sensors of unsaturated FFA status to determine whether FFA are to be stored or oxidized. FFA may regulate lipid metabolism at the level of protein expression or degradation, or via gene transcription and mRNA decay [185]. Nuclear factors that regulate lipogenesis and the storage of FFA include the liver X receptor (LXR) and the sterol regulatory element-binding protein (SREBP) pathways. Transcriptional regulation through SREBP control de novo lipogenesis and lipoprotein uptake mediating cholesterol homeostasis [184]. The LXRs alpha and beta are also cholesterol metabolism and biosynthesis sensors, which result in induced expression of lipogenic enzymes upon stimulation [186]. LXRs, along with the farnesoid X receptor (FXR), also promote storage and catabolism of sterols and their metabolites to prevent excess intracellular cholesterol accumulation [184]. Upon stimulation, the LXRs, which are present in vascular smooth muscle tissue, may lead to foam cell formation, that, in turn, contributes to cardiovascular disease via upregulation of lipogenic genes such as SREBP and fatty acid synthase (FAS) [187].
Three PPAR isoforms exist namely, PPAR alpha, gamma, and delta [188]. These nuclear receptors are activated by natural FFA and lipid-derived ligands. PPAR gamma, which also activates lipogenic enzymes, may exacerbate steatosis and is transcriptionally upregulated in hepatic steatosis [189]. Activation of PPAR gamma induces TG storage in tissues in which it is expressed, including adipose tissue and the intestine [188]. Balancing out the lipogenic and TG storage capacities of PPAR gamma, LXR, and SREBP are the other members of the PPAR family of nuclear receptors, which are
Chapter 11. Lipoprotein Metabolism in Insulin-Resistant States |
219 |
responsible for stimulating the oxidation of FFA. PPAR alpha is highly expressed in liver, heart, and to a lesser extent, in skeletal muscle, and can prevent steatosis upon activation through amplified FFA oxidation processes [188, 189]. In cellular responses to fasting and starvation, PPAR alpha-medi- ated FFA oxidation becomes important for energy metabolism [190, 191]. PPAR delta is ubiquitous, and may also augment FFA oxidation in tissues where PPAR alpha is less abundant [188]. The PPARs may also contribute to the amplification loop of the LXR-mediated cellular efflux of cholesterol, via the ATP-binding cassette (ABC) transporters, ABCA1 and ABCG1, further contributing to their antiatherogenic properties [186]. The balance and interactions of dietary factors via these nuclear receptors determines the partitioning of fatty acids between storage and oxidation.
Nuclear receptor signaling and lipid metabolism are tightly intertwined with insulin signaling and glucose/energy regulation. The IRS downstream of the IR have specific regulatory roles in liver function. IRS-1 is closely linked to glucose homeostasis, whereas IRS-2 is linked to lipid metabolism via downstream metabolic nuclear receptors [192]. Examples of these molecular relationships are illustrated by studies using PPAR alpha KO mice. As expected, PPAR alpha KO mice exhibit markedly reduced FFA oxidation, but are also hyperinsulinemic and hypoglycemic in the fasting state [191].
The pharmacologic ligands of PPAR alpha (fibrates) and PPAR gamma (thiazolidinediones) have been quite successfully developed [188, 193–195]. Fibrates have been found to play a role on insulin sensitivity, via decreases in circulating FFA and TG, and decreases in ectopic lipids deposited in nonadipose tissues [196]. In addition to insulin sensitization of muscle tissue, thiazolidinedione treatment is accompanied by enhanced total body fat oxidation, increases in insulin-stimulated glucose uptake, and decreased plasma FFAs [193]. Thiazolidinediones have also been used successfully in the treatment of nonalcoholic steatohepatitis (NASH) [197]. It appears that thiazolidinedione action on muscle insulin sensitivity may be the result of the secretion of insulin-sensitizing hormones such as adiponectin by adipocytes, and to the lowering of circulating lipids—all events that result in normalized glucose utilization [188, 189, 195]. In addition, another target gene of PPAR gamma is LXR alpha, which is also known for its lipid storage and glucose clearance capacity [186].
A new category of drug therapy for lipid disorders, insulin resistance, and NASH includes PPAR delta agonists. Functions of PPAR delta are less known, but these agonists may play a beneficial role in the treatment of lipid disorders, in particular obesity [194, 195]. PPAR delta can upregulate PGC-1alpha and regulate FFA oxidation in several tissues, which may prove beneficial in the treatment of insulin resistance and obesity [179, 194]. Combination products are also currently being developed for clinical use, targeting both PPAR alpha and PPAR gamma [198].