

биохимия атеросклероза
.pdf118 Kathryn E. Naus and Cheryl L. Wellington
159.Dove DE, Linton MF, Fazio S: ApoE-mediated cholesterol efflux from macrophages: separation of autocrine and paracrine effects. Am J Physiol Cell Physiol 288: C586–C592, 2005.
160.Zhang W-Y, Gaynor PM, Kruth HS: Apolipoprotein E produced by human monocyte-derived macrophages mediates cholesterol efflux that occurs in the absence of added cholesterol acceptors. J Biol Chem 271: 28641–28646, 1996.
161.Kruth HS, Skarlatos SI, Gaynor PM, Gamble W: Production of cholesterolenriched nascent high density lipoproteins by human monocyte-derived macrophages is a mechanism that contributes to macrophage cholesterol efflux. J Biol Chem 269: 24511–24518, 1994.
162.Curtiss LK, Boisvert WA: Apolipoprotein E and atherosclerosis. Curr Opin Lipidol 11: 243–251, 2000.
163.Linton MF, Fazio S: Macrophages, inflammation, and atherosclerosis. Int J Obesity 27: 535–540, 2003.
164.Li AC, Glass CK: The macrophage foam cell as a target for therapeutic intervention. Nat Med 8: 1235–1242, 2002.
165.Stannard AK, Riddell DR, Sacre SM, Tagalakis AD, Langer C, von Eckardstein A, Cullen P, Athanasopouos T, Dickson G, Owen JS: Cell-derived apolipoprotein E (apoE) particles inhibit vascular cell adhesion molecule-1 (VCAM) expression in human endothelial cells. J Biol Chem 246: 46011–46016, 2001.
166.Sacre SM, Stannard AK, Owen JS: Apolipoprotein E isoforms differentially induce nitric oxide production in endothelial cells. FEBS Lett 540: 181–187, 2003.
167.Kothapalli D, Fuki I, Ali K, Stewart SA, Zhao L, Yahil R, Kwiatkowski D, Hawthorne EA, FitzGerald GA, Phillips MC, Lund-Katz S, Puré E, Rader DJ, Assoian RK: Antimitogenic effects of HDL and apoE mediated by Cox-2 dependent IP activation. J Clin Invest 113: 609–618, 2004.
168.Miyata M, Smith JD: Apolipoprotein E alle-specific antioxidant activity and effects on cytotoxicity by oxidative insults and beta-amyloid peptides. Nat Genet 14: 55–61, 1996.
169.Kelly ME, Clay MA, Mistry MJ, Hsieh-Li HM, Harmony JA: Apolipoprotein E inhibition of mitogen-activated T-lymphocytes: production of interleukin 2 with reduced biological activity. Cell Immunol 159: 124–139, 1994.
170.Mistry MJ, Clay MA, Kelly ME, Steiner MA, Harmony JA: Apolipoprotein E restricts interleukin-dependent T-lymphocyte proliferation at the G1A/G1B boundary. Cell Immunol 160: 14–23, 1995.
171.Ishigami M, Swertferger DK, Granholm NA, Hui DY: Apolipoprotein E inhibits platelet-derived growth factor-induced vascular smooth muscle cell migration and proliferation by suppressing signal transduction and preventing cell entry to G1 phase. J Biol Chem 273: 20156–20161, 1998.
172.Swertfeger DK, Hui DY: Apolipoprotein E receptor binding versus heparan sulfate proteoglycan binding in its regulation of smooth muscle cell migration and proliferation. J Biol Chem 276: 25043–25048, 2001.
173.Riddell DR, Vinogradov DV, Stannard AK, Chadwick N, Owen JS: Identification and characterisation of LRP8 (apoER2) in human blood platelets. J Lipid Res 40: 1925–1930, 1999.
174.Riddell DR, Graham A, Owen JS: Apolipoprotein E inhibits platelet aggregation through the L-arginine:nitric oxide pathway. Implications for vascular disease. J Biol Chem 272: 89–95, 1997.
Chapter 6. ABC Transporters and Apolipoprotein E |
119 |
175.Linton MF, Fazio S: Macrophages, lipoprotein metabolism, and atherosclerosis: insights from murine bone marrow transplantation studies. Curr Opin Lipidol 10: 97–105, 1999.
176.Bellosta S, Mahley RW, Sanan DA, Murata J, Newland DL, Taylor JM, Pitas RE: Macrophage-specific expression of human apolipoprotein E reduces atherosclerosis in hypercholesterolemic apolipoprotein E-null mice. J Clin Invest 96: 2170–2179, 1995.
177.Boisvert WA, Spangenberg J, Curtiss LK: Treatment of severe hypercholesterolemia in apolipoprotein E-deficient mice by bone marrow transplantation in mice. J Clin Invest 96: 1118–1124, 1995.
178.Linton MF, Atkinson JB, Fazio S: Prevention of atherosclerosis in apolipoprotein E-deficient mice by bone marrow transplantation. Science 267: 1034–1037, 1995.
179.Zhu Y, Bellosta S, Langer C, Bernini F, Pitas RE, Mahley RW, Assmann G, von Eckardstein A: Low-dose expression of a human apolipoprotein E transgene in macrophages restores cholesterol efflux capacity of apolipoprotein E-deficient mouse plasma. Proc Natl Acad Sci USA 95: 7585–7590, 1998.
180.Van Eck M, Herijgers N, Van Dijk KW, Havekes LM, Hofker MH, Groot PHE, Van Berkel TJ: Effect of macrophage-derived mouse apoE, human apoE3Leiden, and human apoE2 (Arg158-Cys) on cholesterol levels and atherosclerosis in apoE-deficient mice. Arteriosclerosis 20: 119–127, 2000.
181.Van Eck M, Herijgers N, Van Dijk KW, Havekes LM, Hofker MH, Groot PH,
Van Berkel TJ: Effect of macrophage-derived mouse ApoE, human ApoE3Leiden, and human ApoE2 (Arg158→Cys) on cholesterol levels and atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol 20: 119–127, 2000.
182.Van Eck M, Herijgers N, Yates J, Pearce NJ, Hoogerbrugge PM, Groot PH, Van Berkel TJ: Bone marrow transplantation in apolipoprotein E-deficient mice. Effect of ApoE gene dosage on serum lipid concentrations, (beta)VLDL catabolism, and atherosclerosis. Arterioscler Thromb Vasc Biol 17: 3117–3126, 1997.
183.Fazio S, Babaev VR, Murray AB, Hasty AH, Carter KJ, Gleaves LA, Atkinson JB, Linton MF: Increased atherosclerosis in mice reconstituted with apolipoprotein E null macrophages. Proc Natl Acad Sci USA 94: 4647–4652, 1997.
184.Boisvert WA, Curtiss LK: Elimination of macrophage-specific apolipoprotein E reduces diet-induced atherosclerosis in C57Bl/6J male mice. J Lipid Res 40: 806–813, 1999.
185.Van Eck M, Herijgers N, Vidgeon-Hart M, Pearce NJ, Hoogerbrugge PM, Groot PHE, Van Berkel TJ: Accelerated atherosclerosis in C57Bl/6 mice transplanted with ApoE-deficient bone marrow. Atherosclerosis 150: 71–80, 2000.
186.Ishibashi S, Nobuhiro Y, Shimano H, Mori N, Mokuno H, Gotohda T, Kawakami M, Murata J: Apolipoprotein E and lipoprotein lipase secreted from human monocyte-derived macrophages modulate very low density lipoprotein uptake. J Biol Chem 265: 3040–3047, 1990.
187.Haghpassand M, Bourassa P-AK, Francone OL, Aiello RJ: Monocyte/ macrophage expression of ABCA1 has minimal contribution to plasma HDL levels. J Clin Invest 108: 1315–1320, 2001.
188.Aiello RJ, Brees D, Bourassa P-AK, Royer L, Lindsey S, Coskran T, Haghpassand M, Francone O: Increased atherosclerosis in hyperlipidemic mice with inactivation of ABCA1 in macrophages. Arterioscler Thromb Vasc Biol 22: 630–637, 2002.
189.Van Eck M, Bos ST, Kaminski WE, Orsó E, Rothe G, Twisk J, Van Amersfoort ES, Christiansen-Weber TA, Fung-Leung WP, van Berkel TJC, Schmitz G:
120 Kathryn E. Naus and Cheryl L. Wellington
Leukocyte ABCA1 controls susceptibility to atherosclerosis and macrophage recruitment into tissues. Proc Natl Acad Sci USA 99: 6298–6303, 2002.
190.Joyce CW, Amar MJA, Lambert G, Vaisman BL, Paigen B, Najib-Fruchart J, Hoyt RF Jr Neufeld ED, Remaley AT, Fredrickson DS, Brewer HB Jr: The ATP binding cassette transporter A1 (ABCA1) modulates the development of aortic atherosclerosis in C57BL/6 and apoE-knockout mice. Proc Natl Acad Sci USA 99: 407–412, 2002.
191.von Eckardstein A, Langer C, Engel T, Schaukal I, Cignarella A, Reinhardt J, Lorkowski S, Li Z, Zhou X, Cullen P, Assmann G: ATP binding cassette transporter ABCA1 modulates the secretion of apolipoprotein E from human mono- cyte-derived macrophages. FASEB J 15: 1555–1561, 2001.
192.Koo C, Innerarity TL, Mahley RW: Obligatory role of cholesterol and apolipoprotein E in the formation of large cholesterol-enriched and receptoractive high density lipoproteins. J Biol Chem 260: 11934–11943, 1985.
193.Huang ZH, Lin CY, Oram JF, Mazzone T: Sterol efflux mediated by endogenous macrophage apoE expression is independent of ABCA1. Arterioscler Thromb Vasc Biol 21: 2019–2025, 2001.
194.Huang ZH, Mazzone T: ApoE-dependent sterol efflux from macrophages is modulated by scavenger receptor class B type I expression. J Lipid Res 43: 375–382, 2002.
Biochemistry of Atherosclerosis edited by S.K. Cheema, Springer, New York, 2006
7
Provision of Lipids for Very Low-Density Lipoprotein Assembly
DEAN GILHAM AND RICHARD LEHNER
Abstract
Atherosclerosis is the principal cause of heart attack and stroke in the Western world. The relationship between high levels of low-density lipoprotein (LDL) and atherosclerosis has been known for several decades. LDL is derived from very low-density lipoprotein (VLDL) in the blood via a complex series of reactions involving hydrolases and transfer of lipids and apoproteins among the circulating lipoproteins. Hepatic VLDL assembly and secretion is dependent on lipid availability. Thus stimulation of hepatic triacylglycerol (TG), phospholipid, and cholesterol synthesis increases the secretion of VLDL while inhibition of lipid synthesis has the opposite effect. Several lines of evidence suggest that the assembly of VLDL takes place in two or more steps. The first step involves the generation of dense nascent lipoprotein particles in the endoplasmic reticulum by cotranslational lipidation of apolipoprotein B and these particles mature into a VLDL by posttranslational addition of core lipids such as TG and cholesteryl ester (CE). De novo synthesized TG accounts for only a minor fraction of the TG secreted with VLDL. The majority (60–70%) of VLDL-TG is derived from lipolysis of stored TG to partial acylglycerols and fatty acids followed by reesterification by the ER-localized acyltransferases. Recently, the genes of a number of enzymes involved in the provision of lipids for VLDL assembly have been cloned and in some cases mice in which these genes were ablated have been obtained. This chapter discusses recent advances in VLDL assembly with emphasis on the lessons learned from genetically modified mouse models.
Keywords: acyltransferase; lipolysis; liver; triacylglycerol; triacylglycerol hydrolysis; VLDL
Abbreviations: ACAT, acyl coenzyme A:cholesterol acyltransferase; AGPAT, 1-acylglycerol-3-phosphate acyltransferase; apo, apolipoprotein; CCT, cytidine triphosphate:phosphocholine cytidylyltransferase; CHAPS, [(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate; CE, cholesteryl ester; CVD, cardiovascular disease; DG, diacylglycerol; DGAT, diacylglycerol acyltransferase; DGTA, diacylglycerol transacylase; E600, diethyl-p-nitrophenyl phosphate; ER; endoplasmic reticulum; GPAT, glycerol-3-phosphate acyltransferase; HDL, high-density lipoprotein; HSL, hor- mone-sensitive lipase; LDL, low-density lipoprotein; MG, monoacylglycerol; MGAT, monoacylglycerol acyltransferase; MTP, microsomal triglyceride transfer protein; PC, phosphatidylcholine; PEMT, phosphatidylethanolamine N-methyltransferase; PAPH,
121
122 Dean Gilham and Richard Lehner
phosphatidic acid phosphohydrolase; PPAR, peroxisomal proliferator-activated receptor; SCD, stearoyl-CoA desaturase; SREBP, sterol regulatory elements binding protein; TG, triacylglycerol; TGH, triacylglycerol hydrolase; VLDL, very low-density lipoprotein.
Triacylglycerol Metabolism is Linked to Several Human Diseases
The ability to store neutral lipids plays a critical role in the ability of an organism to withstand fuel deprivation. Triacylglycerol (TG) is the most concentrated form of energy available to biological tissues. It can provide over double the amount of energy by metabolic oxidation than an equal mass of either carbohydrates or proteins [1]. TG is composed of a glycerol backbone to which three fatty acids are attached via ester bonds. Most cells and tissues have the ability to synthesize and store TG to some degree. TG metabolism is of tremendous clinical relevance. Dysregulation of TG lipid synthesis, transport, or storage contributes to the development of disease. Consequently there is tremendous clinical and pharmacological relevance for enzymes that metabolize TG. Obesity alone is associated with type 2 diabetes, hyperlipidemia, cardiovascular disease, stroke, gallbladder disease, and some cancers [2–7]. Excessive TG storage within muscle and pancreatic β-cells correlates with insulin resistance, leading ultimately to diabetes [8, 9]. Excessive hepatic TG secretion contributes to high levels of circulating TG in the blood, which is a risk factor for cardiovascular disease (CVD). Plasma TG levels >150 mg/dL or 1.70 mM in a fasted state are considered elevated [10].
Components and Classification of Lipoproteins
Neutral lipids such as TG and CE are hydrophobic in nature, and hence have very low solubility in aqueous environments including blood. Therefore, in order to be transported throughout the body, where they are required for various cellular functions, hydrophobic lipids are packaged into complexes with specific proteins known as apolipoproteins (apos) as well as partly polar phospholipids. All mature TG-rich lipoproteins consist of a hydrophobic core of nonpolar lipids including cholesteryl esters (CE) and TG surrounded by a phospholipid monolayer. Free or unesterified cholesterol can also exist within the monolayer of phospholipid. Apos are also amphipathic and confer solubility to the lipoprotein particle as well as influence trafficking via interactions with various cell surface receptors, hydrolytic enzymes, and lipidtransferring proteins [11]. Most apos are exchangeable; they can detach from the surface of a lipoprotein then reattach to another. Hence the complement
Chapter 7. Provision of Lipids for VLDL Assembly |
123 |
of apos changes during the lifetime of these particles. The exception to apos that exchange is more hydrophobic and very large apoB, which is integral and remains bound to the particle it is initially assembled with [12, 13].
Lipoproteins are classified based on their density, which is determined by their lipid and protein composition and is inversely proportional to their diameter. Chylomicrons are the largest and least dense class of TG-rich lipoproteins, with a very TG-rich core (75–1200 nm in diameter) [14]. They are synthesized by the small intestine utilizing ingested dietary lipids, and are carried by the lymphatic system to the bloodstream. Very low-density lipoprotein (VLDL) is the second largest lipoprotein (30–100 nm), and is synthesized and secreted by the liver via an intricate secretory pathway [15, 16]. VLDL is also rich in TG and is catabolized in the circulatory system via a complex series of reactions involving lipases and the transfer of lipids and apos among lipoproteins [17] to produce intermediate (IDL) and low-density lipoproteins (LDL; LDL is approximately 18–25 nm in diameter).
The final class is the smallest (25 nm in diameter or smaller), and these particles are known as high-density lipoproteins (HDL). HDL does not carry apoB, but rather contains at least one molecule of apoA-I among other apos [11]. HDL has a relatively CE-rich core. It is important for the removal of excess cholesterol from peripheral (nonhepatic) cells and its transport back to the liver for recycling or excretion as bile. The collection of cholesterol from the periphery and delivery back to the liver is known as the reverse cholesterol transport pathway. The cardioprotective nature of HDL has been shown in numerous epidemiological studies worldwide over the last 50 years. A striking inverse correlation between HDL cholesterol and incidence of CVD has been observed [18–21], strongly suggesting that a major risk factor for CVD development is low-plasma HDL [22, 23]. Although the detailed mechanisms of how cells efflux cholesterol to HDL and how HDL protects against atherosclerosis are not fully elucidated, potential strategies to treat and prevent CVD exist in raising plasma HDL levels [24].
VLDL Assembly and Secretion
The major apo component of chylomicrons, VLDL, and the sole apo of LDL is a single apoB molecule per particle [12, 17]. Several excellent reviews regarding the mechanism of VLDL assembly and secretion have been published [16, 25, 26]. Gene targeting experiments in mice revealed that apoB is the sole apo required for the secretion of a VLDL particle [27]. ApoB is encoded by a single gene, though it occurs in two forms due to mRNA editing by a deaminase that converts codon 2153 from CAA (glutamine) to a stop codon [28, 29]. The larger form, termed apoB100, is the sole form of apoB secreted by the human liver and includes 100% of the mature protein. Human apoB100 has 4536 amino acids and a calculated molecular weight of over 512
124 Dean Gilham and Richard Lehner
kDa [30, 31]. The shorter form, apoB48, represents the N-terminal 48% of apoB100 with 2152 amino acids and a calculated molecular weight of over 264 kDa [13]. ApoB48 is secreted by human intestine on chylomicrons for transport of dietary lipids. While apoB100 is capable of an interaction with the LDL receptor to mediate cellular uptake via endocytosis, apoB48 lacks the LDL-receptor interacting domain. It is initially counterintuitive that chylomicrons can be up to ten times larger than VLDL and carry more lipids, while at the same time they contain the shorter apoB protein. This means that the C-terminal 52% of apoB100 is not necessary for acquisition of large amounts of lipid during lipoprotein assembly. Both forms of apoB are secreted by the liver of rats and mice [27], making these animals a more challenging model in which to investigate lipoprotein metabolism as it applies to humans.
Triacylglycerol Biosynthesis
The fatty acids present within the TG molecule are provided by dietary lipids or are generated de novo. Variations in the composition and quantity of the diet affect the rate of de novo fatty acid and TG synthesis to satisfy the body’s demands for lipid (or energy) storage [32]. The source of the fatty acids for TG synthesis varies depending upon the tissue [33]. For example, the enterocyte takes up dietary fatty acids and 2-monacylglycerols (MG) and resynthesizes TG from these substrates primarily through the 2-monoacylglycerol (MG) pathway. Hepatocytes take up fatty acids derived from remnant lipoproteins and free fatty acids released from adipocytes. It is generally accepted that the synthesis of TG is controlled primarily by the amount of fatty acid available. Thus, de novo TG synthesis is enhanced only when fatty acid synthesis or uptake exceeds the cellular need for energy and phospholipid synthesis. In hepatocytes, as well as adipocytes, de novo TG synthesis is primarily achieved through the glycerol-3-phosphate pathway (Fig. 7.1).
The Glycerol-3-Phosphate Pathway of TG Biosynthesis
The glycerol-3-phosphate pathway is the main route of TG biosynthesis in nonenteric tissues. Briefly, it begins with the acylation of glycerol-3-phosphate by glycerol-3-phosphate acyltransferase (GPAT) at the sn-1 position to form 1-acylglycerol-3-phosphate. Subsequently, the acylation of 1-acylglycerol-3- phosphate at the sn-2 position by 1-acylglycerol-3-phosphate acyltransferase (AGPAT) (also referred to as lysophosphatidic acid acyltransferase) synthesizes phosphatidic acid (PA). The hydrolysis of PA by phosphatidate phosphohydrolase (PAPH) generates sn-1,2-diacylglycerol (DG). DG is then acylated to form TG via the action of diacylglycerol acyltransferase (DGAT). The utilization of molecular cloning and gene targeting techniques has elucidated many new insights regarding these enzymes and their regulation. The
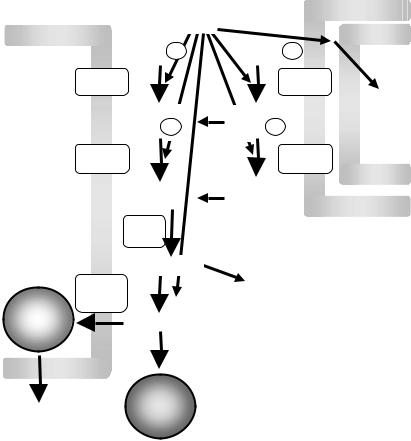
Chapter 7. Provision of Lipids for VLDL Assembly |
125 |
ER membrane |
Fatty acyl-CoA |
|
||
|
|
|
||
|
Glycerol-3- P |
Glycerol-3- P |
|
|
|
GPAT |
|
GPAT |
|
|
|
1-Acylglycerol - |
1-Acylglycerol- |
β-Oxid’ation |
|
|
3- P |
3- P |
|
|
AGPAT |
AGPAT |
||
ER |
|
Phosphatidic |
Phosphatidic |
|
lumen |
|
|
||
|
acid |
acid |
|
|
|
|
PAPH |
|
Mitochondrial |
|
|
|
|
membranes |
|
|
Diacylglycerol |
|
|
|
DGAT |
|
Phospholipid |
|
|
|
synthesis |
|
|
|
DGTA |
|
|
|
|
|
|
|
|
VLDL |
|
Triacylglycerol |
|
|
storage
secretory droplet pathway
FIGURE 7.1. The glycerol phosphate pathway of triacylglycerol synthesis. Glycerol-3- phosphate is acylated to form 1-acyl-glycerol-3-phosphate by glycerol-3-phosphate acyltransferase (GPAT). A subsequent acylation is accomplished by 1-acyl-glycerol-3- phosphate acyltransferase (AGPAT), resulting in phosphatidic acid. Hydrolysis of the sn-3-phosphate through the action of phosphatidic acid phosphohydrolase (PAPH) produces diacylglycerol. Diacylglycerol may be converted to triacylglycerol by diacylglycerol acyltransferase (DGAT) or diacylglycerol transacylase (DGTA). Alternately, it can be diverted towards phospholipid biosynthesis. In liver, triacylglycerol is stored in droplets or secreted on VLDL.
regulation of TG synthesis and its breakdown determines the quantity of TG stored within a cell.
Glycerol-3-Phosphate Acyltransferase
GPAT represents the committed step for the synthesis of all glycerolipids. Two isoforms for this enzyme exist. A mitochondrial GPAT has been cloned
126 Dean Gilham and Richard Lehner
and demonstrated to be tightly regulated [34]. A microsomal GPAT activity also exists, though a mammalian cDNA has not been cloned and its activity appears to be largely unregulated [34]. The murine mitochondrial GPAT cDNA is primarily expressed in liver, adipose, and muscle tissues [35].
The nutritional and hormonal regulation of the murine mitochondrial GPAT is well characterized and occurs primarily at the transcriptional level. Posttranslational reduction of mitochondrial GPAT activity has been reported to occur upon stimulation of adenosine monophosphate-activated kinase, a sensor of cellular fuel deprivation [36].
GPAT activity has strong links to obesity. For example, Zucker rats are a commonly used model for obesity as well as insulin resistance. These animals exhibit enhanced GPAT activity in both hepatic and adipose tissues [37]. The regulation of mitochondrial GPAT is consistent with a function in reducing TG synthesis and increasing oxidation when nutrients are limiting, and conversely to direct fatty acids toward TG synthesis and away from oxidation when energy is in excess [38]. The localization of this GPAT activity on the cytosolic surface of mitochondria also suggests a role in preventing fatty acids (more accurately fatty acyl-CoAs) from entering this organelle and undergoing β-oxi- dation under conditions of metabolic excess by converting it to 1-acylglycerol- 3-phosphate and thus directing the fatty acid towards TG synthesis (Fig. 7.1).
Acylglycerol-3-Phosphate Acyltransferase
This enzyme is also referred to as lysophosphatidic acid acyltransferase [34]. Several AGPAT cDNA sequences have been identified. Human and mouse cDNAs of 2 and 1.8 kb, respectively that encode proteins with AGPAT activity were identified and designated AGPAT-1 [39, 40]. 3T3-L1 cells are a fibroblastic preadipocyte cell line that can be differentiated into cells with an adipocyte phenotype. AGPAT-1 overexpression in 3T3-L1 adipocytes inhibited fatty acid release while promoting its uptake and TG storage [41]. Expression of AGPAT-1 mRNA has been observed in all tissues examined, with highest expression in skeletal muscle [39, 40]. AGPAT-2 mRNA is expressed at highest levels in the liver, heart, and adipose tissues, and much less in other tissues [42, 43]. The protein encoded by the AGPAT-2 cDNA shares 48% homology with AGPAT-1 and is expressed at twofold higher levels than AGPAT-1 in the adipose tissue, similar levels in liver and AGPAT-1 is expressed at 1.8-fold greater levels than AGPAT-2 in skeletal muscle [44]. AGPAT-1 and -2 were localized to the endoplasmic reticulum (ER) membrane [45, 46]. Several individuals with congenital generalized lipodystrophy were discovered to have a variety of different mutations in the AGPAT-2 gene that led to the production of a dysfunctional protein [44]. These observations suggest that AGPAT-2 is necessary for TG synthesis in adipocytes. Three additional AGPAT cDNAs have been designated AGPAT-3, -4, and -5, respectively. The corresponding mRNAs are barely detectable in liver, adipose, and skeletal muscle and do not yet have a clearly identified function [44].
Chapter 7. Provision of Lipids for VLDL Assembly |
127 |
Phosphatidic Acid Phosphohydrolase
Two forms of phosphatidic acid phosphohydrolase (PAPH) activity exist, PAPH-1 and PAPH-2 (also called lipid phosphate phosphohydrolase). Several cDNAs for the PAPH-2 family or lipid phosphate phosphohydrolases have been cloned. These enzymes appear to play a role in signal transduction pathways by generating the lipid second messenger, DG [47]. PAPH-1 activity exists in both cytosol and microsomal fractions. It has been proposed that long-chain fatty acids stimulate the translocation of PAPH-1 from the cytosol to the ER membrane where PA is hydrolyzed to DG [48, 49]. A cDNA encoding an enzyme with PAPH-1 activity has not been cloned, but this activity is considered as the rate-limiting step for de novo TG synthesis. There is a strong correlation between the activity of membrane-associated PAPH-1 and the rate of TG synthesis [50]. PAPH-1 activity is subjected to dietaryand hormone-induced changes [50]. PAPH-1 activity was increased 2–3-fold by glucagon, cyclic adenosine monophosphate, and dexamethasone in primary rat hepatocytes [51–53]. It was hypothesized that enhanced PAPH-1 activity enables the liver to increase its ability to synthesize TG during the period of maximum feeding.
Diacylglycerol Acyltransferase
DGAT catalyzes the final step in TG synthesis. Two DGAT cDNAs have been cloned. DGAT-1 is ubiquitously expressed in human and murine tissues, with the highest levels of expression in the small intestine, colon, testis, thymus, heart, and skeletal muscle [54]. The mammalian DGAT-2 cDNA is unrelated to the mammalian DGAT-1 cDNA and was cloned based on its homology with a fungal DGAT cDNA [55]. Both proteins have been localized to microsomal membranes. DGAT-2 is highly expressed in liver and adipose, which correlates with a high capacity to synthesize TG in these tissues. Analysis of the predicted amino acid sequence from the DGAT-2 cDNA suggests the presence of a single transmembrane segment and a domain with homology to the glycerol-binding domain of GPAT and AGPAT. The glycerol-binding domain of DGAT-2 is predicted to be within the ER lumen where the synthesis of TG would be ideally linked to the site of VLDL assembly. There is an experimental evidence for the existence of overt (cytosolic) and latent (lumenal) DGAT activities in microsomes [56]. Hence, different acyltransferases (or transacylases), acting in different subcellular compartments could be involved in the synthesis of TG, and the extent of their respective activities could channel TG preferentially for storage within a cytosolic storage pool or towards the secretory pathway via assembly into lipoproteins. Whether the DGAT-1 or -2 enzyme is responsible for directing TG towards VLDL versus storage has been investigated.
Recently, adenovirus constructs were used to overexpress DGAT-1 or DGAT-2 in mice [57]. Intriguingly the data showed that DGAT-1 overexpression led to increased VLDL, while DGAT-2 overexpression led to TG