

Загальні клінічні прояви та біохімічні показники
Більшість метаболічних хвороб яскраво проявляються в неонатальному періоді після припинення плацентарного кровотоку і початку функціонування власного метаболізму. Але оскільки МХ реалізується ще внутрішньоутробно, результати порушеного метаболізму плоду можуть вплинути на стан здоров’я вагітної і тому акушерський анамнез грає величезну роль в антенатальному пошуку МХ:
спонтанний аборт або мертвонародження в анамнезі повинні розцінюватися як елімінація нежиттєздатної дитини. Чоловіча стать такого плоду може свідчити про Х - зчеплену форму МХ;
наявність патологічних змін у вагітної, таких як затяжна блювота вагітних або гостра жирова дистрофія печінки, можуть бути наслідком порушення у плоду окислювання жирних кислот.
Мати, яка є гетерозиготним носієм патологічного гену, переживає метаболічний стрес внаслідок впливу продуктів метаболізму плоду, які потрапляють в її кровоток и порушують стан здоров’я.
Гіперактивність плоду, ритмічні його рухи свідчать ще про один прояв порушеного метаболізму – про судоми, які виникають внутрішньоутробно.
Коли слід думати про спадкові хвороби обміну?
Якщо в неонатальному періоді початковими ознаками є:
летаргія
відмова від їжі
втрата маси
поліпноє
гіпотермія
гіпотонія
незвичні рухи
гепатомегалія.
Якщо в подальшому розвитку хвороби клінічні ознаки характеризуються:
судомами
поліорганними змінами
комою.
Звичайно ці симптоми неспецифічні, а метаболічні хвороби часто сполучаються з інфекцією і характеризуються прогресуючим плином.
В неонатальному періоді всі прояви МХ можна підрозділити на 3 основні групи:
порушення синтезу і розпаду складних молекул;
інтоксикація;
енергетична недостатність.
Порушення синтезу складних молекул звичайно призводить до патології ембріогенезу і, як наслідок, до появи дизморфічних рис обличчя. Наприклад, порушення пероксисомного біогенезу формує фенотип синдрому Цельвегера, Рефсума; генетичний блок в синтезі холестеролу – фенотип синдрому Сміта-Лемлі-Опітца (рис. 1).
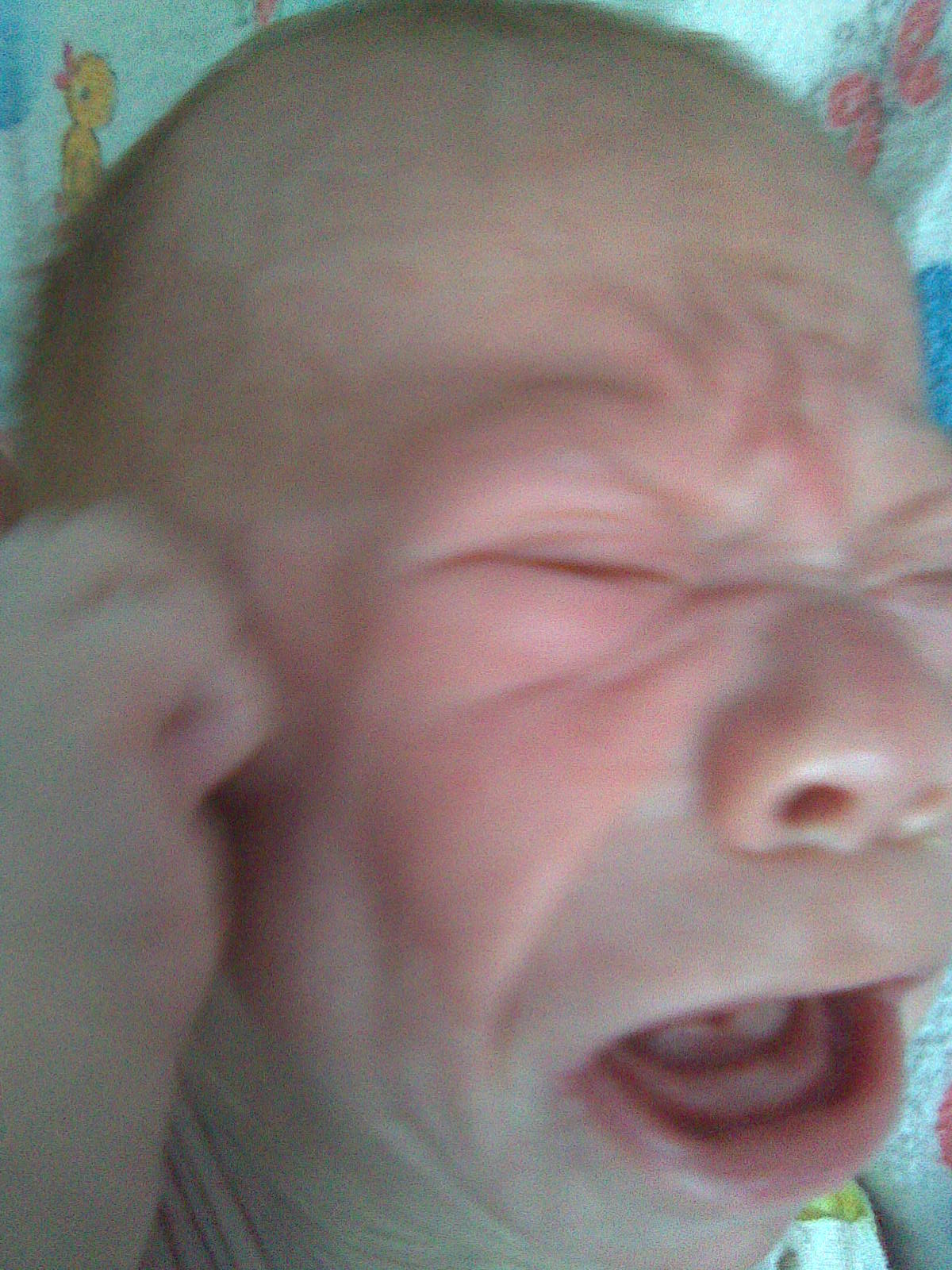
Рис. 1. Дизморфічні риси обличчя у дитини з органічною ацидурією
Порушення розпаду складних молекул призводить до їх накопичення і, внаслідок цього, до формування специфічного зовнішнього вигляду, який дозволяє лікарю-клініцисту запідозрити таке захворювання. Наприклад, синдром Гурлер (наслідок порушення розпаду мукополісахаридів), хвороба Тея-Сакса (порушення розпаду гангліозидів) (рис. 2-4).
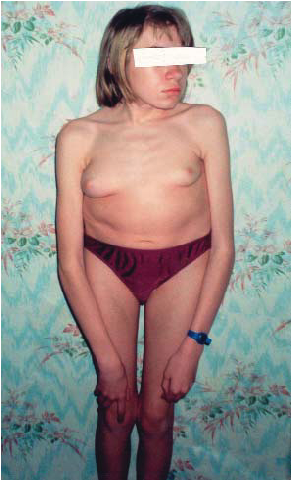
Рис. 2. Мукополіcaхаридоз, IV тип
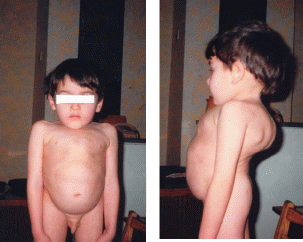
Рис. 3-4. Мукополіcaхаридоз, IV тип
Інтоксикація змушує запідозрити МХ. З першим ковтком молока матері дитина піддається впливу метаболітів і, як наслідок, у неї розвивається енцефалопатія вже у перші 72 години життя. Наприклад, при порушенні циклу сечовини (блок виведення аміаку), органічних ацидуріях (блок в обміні амінокислот), галактоземії (блок в обміні галактози) (рис. 5).
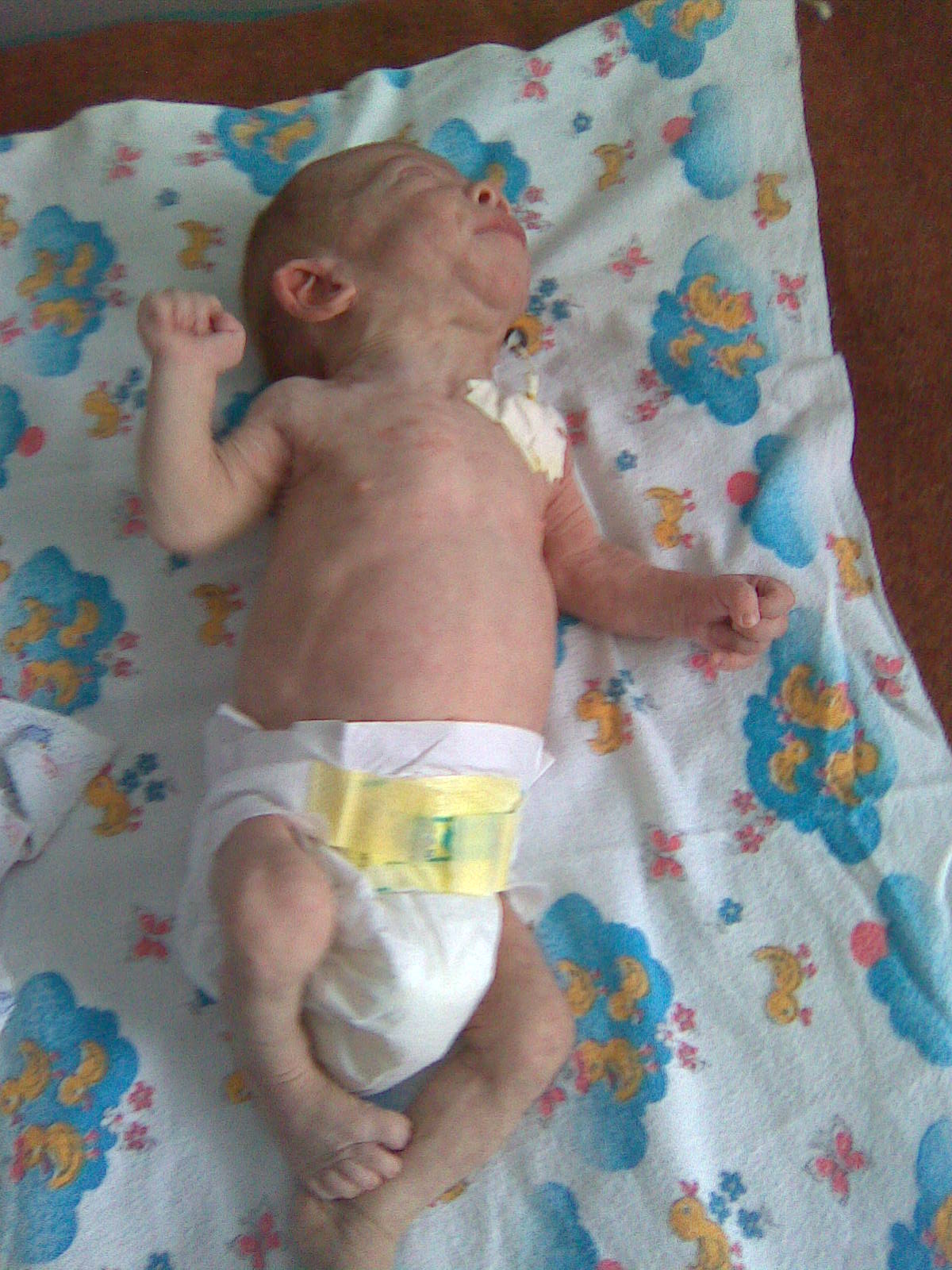
Рис. 5. Прояви енцефалопатії у дитини з органічною ацидурією
Енергетична недостатність.
При порушенні біоенергетичного обміну початкові прояви характеризуються лактат-ацидозом, який відображає порушення продукції АТФ. Спектр клінічних проявів доволі широкий. Частим симптомом є порівняно рання інтоксикація, яка може мати затяжний характер з розвитком декомпенсації. Продукція енергії може також порушуватися, якщо джерело енергії (їжа) не потрапляє до організму в необхідній кількості та якості або розвивається інтеркурентна інфекція (рис. 6).

Рис. 6. Синдром млявої дитини при органічній ацидурії
Дослідження механізму запуску метаболічних кризів при спадкових порушеннях обміну дозволило відмітити ряд факторів, на які вказують батьки як на ініціюючі. Серед них:
- голодування;
- інфекції;
- операції;
- травми.
Ці фактори виявлені у хворих з порушенням білкового, вуглеводного та біоенергетичного обміну.
Встановлено, що високий рівень споживання білків або прискорений білковий катаболізм ініціює проявлення аміноацидопатій, органічних ацидурій, дефекти циклу сечовини; значне вуглеводне навантаження реалізує мітохондріопатії; споживання великої кількості фруктів реалізує нестерпність фруктози; молочних продуктів – галактоземії; значне жирове навантаження призводить до маніфестації порушень окислення жирних кислот; зловживання ліками впливає на маніфестацію порфірії, недостатності глюкозо-6-фосфат-1-дегідрогенази, порушень окислення жирних кислот; зловживання фізичними навантаженнями ініціює гемолітичний криз при прихованій мітохондропатії.
Відмінною властивістю МХ є наявність безсимптомного періоду, який триває різний час у різних хворих і при різних порушеннях обміну. Однак це ствердження відносно ознаки є з самого початку "помилками" обміну, але необхідно навчитися їх бачити й адекватно оцінювати. Цілком очевидно, що досить тривалий час ознаки МХ можуть мати неспецифічний характер, і їх легко сплутати з "сепсисом", "родовою травмою", "перинатальною енцефалопатією". Пізня діагностика МХ призводить до повної маніфестації МХ та інвалідизації, тоді як рання дає змогу проводити своєчасне і ефективне лікування – патогенетичну терапію, яка направляє метаболізм в потрібне русло.
В теперішній час систематизовані загальні клінічні прояви МХ, які дозволяють запідозрити цей вид спадкової патології в різних вікових групах.
До них відносяться:
- невстановлений діагноз; - невстановлена причина раптової смерті; - незвичайний запах повітря, яке видихається, тіла, сечі, колір сечі; - наявність сибсів з невстановленим діагнозом, сепсисом, енцефалопатією; - порушення психофізичного розвитку; - поява ознак хвороби при переході на іншу дієту; - пристрасть або відраза до окремих продуктів; - судоми; - порушення м’язового тонусу; - органомегалія; - змінення шкіри (попрілості, товста шкіра, пігментація); - обмеження рухливості суглобів; - гірсутизм; - гикавка, блювання; - кровноспоріднений шлюб. |
Сучасна класифікація метаболічних хвороб розроблена J.Zschoke, G.Hoffmann (1999) виглядає таким чином (табл.1):
Таблиця 1