

Моносомія 21q
Аномалію описали у вигляді del(21)(q11→qter) Lejeunіз співавт.,1964; Fryns із співавт.,1977; Reynolds із співавт.,1985. Спостерігається генералізована гіпотонія.
Характеристика фенотипу:
Череп: мікроцефалія.
Очі: антимонголоїдний розріз очей, блефарохалазія.
Рот: високе піднебіння, розколина верхньої губи.
Підборіддя: мікрогнатія.
Вуха: великі низько розташовані вушні раковини з аномальними лініями.
Хребет: аномалії.
Шкіра: аномалії нігтів.
Вади внутрішніх органів: кардіопатія, пілоростеноз, агенезія мозолистого тіла.
Кільцева хромосома 21
Аномалію описали у вигляді r(21)(p11.2 і q22.3) Rіcher із співавт.,1977; Cruіsі, Engel, 1986 Hertz, 1987.
Клінічні ознаки аналогічні ознакам синдрому моносомії довгого плеча хромосоми 21.
Трисомія 21q
Аномалія описана у вигляді trі(21)(q11→qter) Hagemeіjer і Smіt,1977; Matteі із співавт., 1981; Delabar із співавт.,1987.
Клінічні прояви аналогічні проявам синдрому Дауна.
Синдром Дауна (трисомія 21)
Описаний Down,1866; Waardenburg,1932 у вигляді trі(21;21q22).

Рис. 49. Синдром Дауна. Жіночий каріотип.
47,ХХ,+21
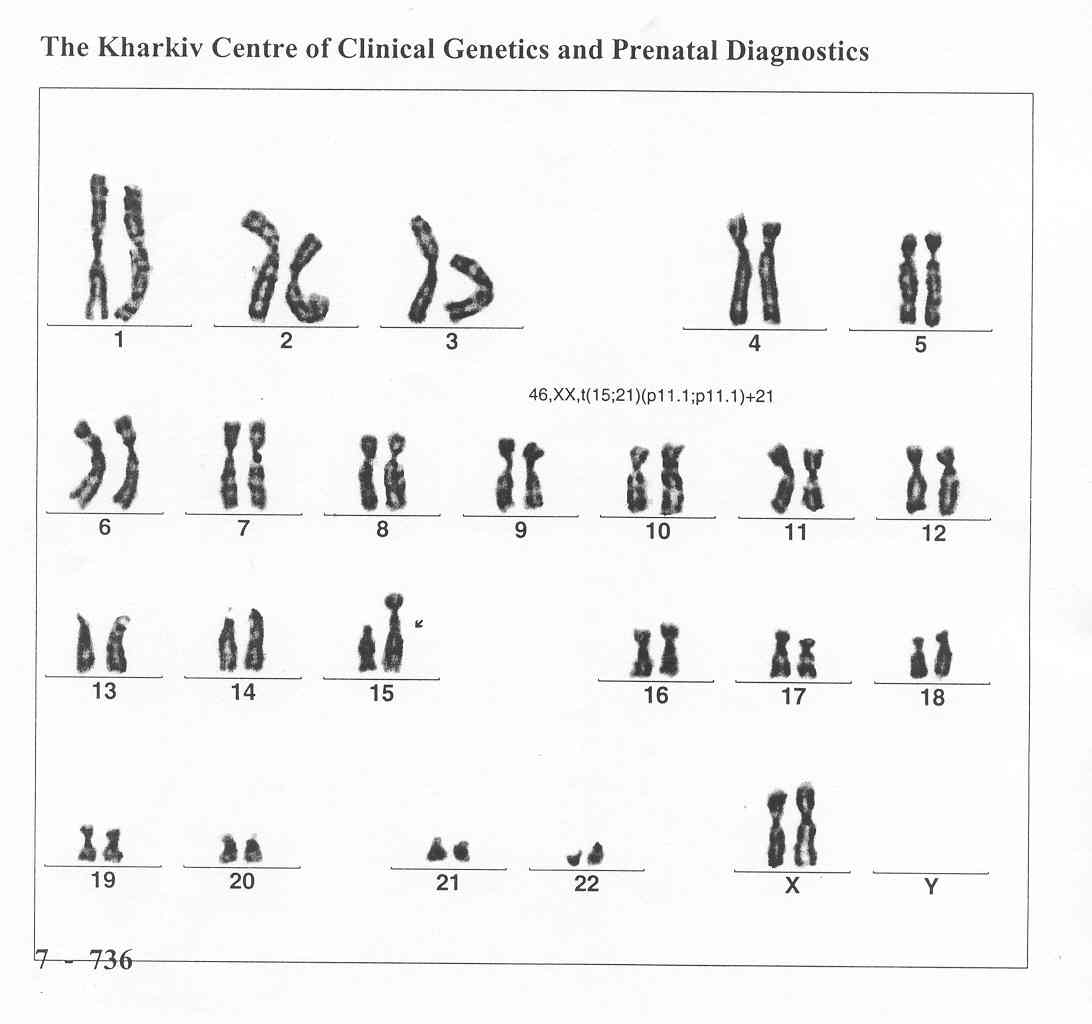
Рис. 50. Транслокаційна форма синдрому Дауна
Жіночий каріопит 45,ХХ,der(15;21)(q10;q10)
Загальні аномалії: гіпотонія, тенденція тримати рот відкритим з висунутим язиком, неперфорований відхідник, гіпермобільність суглобів, відносно невеликий рот, незграбна хода.
ЦНС: розумова відсталість.
Череп і лице: брахіцефалія з відносно плоскою потилицею, тенденція до розташування волосяного завитка на середньотім'яній лінії. Незначна мікроцефалія з монголоїдним розрізом очних щілин. Тонкі кістки черепа з пізнім закриттям тім'ячка. Гіпоплазія або аплазія лобних пазух, коротке тверде піднебіння.
Ніс. Маленький ніс із низьким переніссям і тенденцією до наявності внутрішньої епікантусної складки.
Очі: цяточки на райдужці (плями Брушфільда) з периферічною гіпоплазією райдужки, дрібні кришталики під час огляду за допомогою лампи (59%). Аномалії рефракції.
Вуха: маленькі, низько розташовані з завитком, що перекривається, і протизавитком, що виступає, мочки вух відстовбурчені, маленькі або відсутні.
Рот: губи маленькі, неправильно розташовані, карієс рідше, ніж зазвичай.
Шия: коротка.
Руки: відносно короткі п’ясткові і фаланги. V палець - гіпоплазія середньої фаланги (60%) з клінодактилією (50%) і/або єдина згинальна складка. "Мавпяча" складка (45%).
Дистальний аксіальний трирадіус (84%). Ульнарні петлі на більшості пальців (35%).
Ноги: широка щілина між І і II пальцями.
Таз: гіпоплазія зі зменшеними клубовим і ацетабулярним кутами.
Серце: вади приблизно у 40 % хворих. Дефекти міжпередсердних, міжшлуночкової перегородок, відкрита артеріальна протока, аберантна підключична артерія (вади перелічено в порядку зменшення частоти).
Шкіра: широкі складки в дорсальних відділах шиї. Мармурова шкіра, особливо в ділянці кінцівок. Суха шкіра з явищами гіперкератозу (75 %).
Волосся: тонке, м'яке й часто рідке, пряме волосся на лобку в юнацькому віці.
Геніталії: у чоловіків відносно маленький пеніс. Гіпоспадія в період фертильності проявляється у 100 % немовлят.
Рідкісні фенотипні прояви: косоокість (33 %), ністагм (15 %), кератоконус (6 %), катаракта (1,3 %), низько розташовані вуха, 2 центри скостеніння в рукоятці груднини, лійкоподібна або конічна грудна клітка, трахеостравохідна нориця, атрезія дванадцятипалої кишки, тетрада Фалло, неповне з'єднання хребетних дужок у нижніх відділах хребта (37 %), лише 11 ребер, крипторхізм (27 % від народження до 9 років і 14 % — після 15 років), синдактилія II і III пальців стоп. Частота лейкемії становить 1 : 15 або наближається до 1 %. Порушення функції щитоподібної залози: елементарний зоб або гіпотиреоз. Характерні риси в немовлят: діагноз зазвичай встановлюють невдовзі після народження. Нижче представлено 10 особливостей синдрому Дауна, з яких Хол виявив принаймні 4 у 48 % немовлят із синдромом Дауна та 6 і більше —у 89 %.
Гіпотонія — 80 %. Слабкий рефлекс Моро — 85 %. Гіпермобільність суглобів — 80 %. Плоский профіль лиця — 90 %. Косе розташування очної щілини — 80 %. Аномальні вуха — 60 %. Дисплазія таза — 70 %. Дисплазія середньої фаланги V пальця на кистях — 60 %. "Мавпяча" складка — 45 %.
У 1866 р. англійський лікар Джон Ланґдон Даун уперше описав хворобу, яку згодом назвали його ім'ям. Д. Даун — видатний лікар, який випереджав свій час, описуючи хворих з генетичними або частково генетичними відхиленнями, такими як псевдогіпертрофічна м'язова дистрофія, синдром Прадера-Віллі, синдром Аспержера. Даун назвав цю хворобу "монголоїдною ідіотією" і зробив неадекватний висновок, що хворі із трисомією 21 прогредієнтно деградують. Однак серед своїх сучасників він відзначився тим, що визнав ще одну "людську модель" і виступив проти расистських теорій. Терміни, пов'язані з монголоїдною расою, використовували до 1930-х років і вилучили після подання офіційного протесту монгольським урядом.
У розвиток знань про хворобу Дауна великий внесок зробив Шуттельворс. Обстеживши 350 хворих, він дійшов висновку, що вік матері, як і виснаження репродуктивної функції, що розвивається за повторних вагітностей, є чинниками, відповідальними за вроджені вади розвитку. Спостереження, що діти із хворобою Дауна народжуються в багатодітних родинах останніми, було зроблено в XIX ст. Фразером і Пенроузом, які встановили наявність залежності між віком матері і виникненням синдрому Дауна. Незважаючи на ці досягнення в розумінні епідеміології синдрому Дауна, наприкінці XIX — на початку XX ст. були зроблені неправильні висновки про зв'язок синдрому Дауна із сифілісом у матері, туберкульозом, гіпотиреозом, алкоголізмом батька, порушенням ендокринної функції у плода, дегенерацією статевих клітин, емоційною травмою матері під час гестації. Усі ці чинники діяли відразу або один за одним. Варто віддати належне офтальмологові Варденбург й американському педіатрові Блейру, які висловили думку про хромосомну етіологію хвороби Дауна ще за 25 років до демонстрації маленької акроцентричної хромосоми 21. Лежен у 1959 р. зробив припущення щодо хромосомної етіології синдрому Дауна після того, як Тіо й Леван установили, що нормальна диплоідна кількість хромосом становить 46. Незабаром після цього Полані із співавт. (1960) виявили транслокаційну форму синдрому Дауна в дочки жінки віком 21 рік, яку було обрано для вивчення, оскільки дослідники дійшли висновку, що деякі індивіди можуть уражуватися хворобою якимись окремими, незалежними від віку матері, механізмами. Наступного року (1961) Кларк зі співавт. повідомили про мозаїчний синдром Дауна, продемонструвавши наявність нормального диплоїдного набору і трисомії 21 у шкірі й крові, взятих у дворічної дівчинки з фенотипними ознаками синдрому Дауна, але з практично нормальним розумовим розвитком.
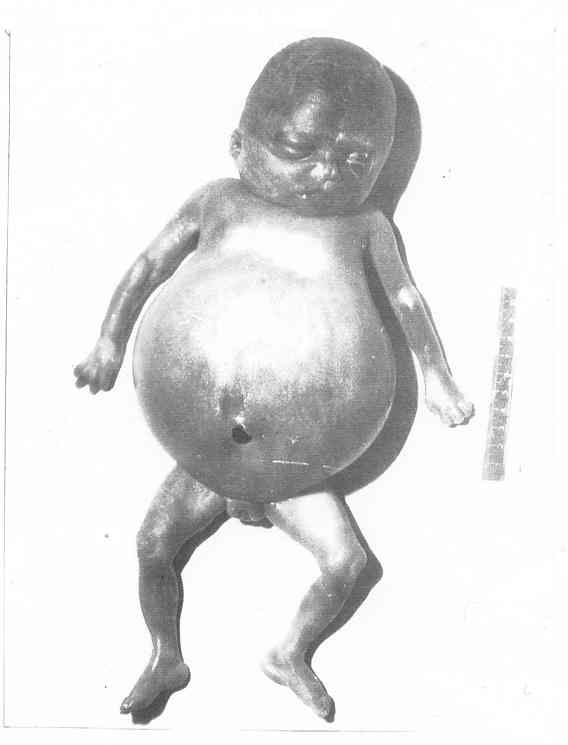
Рис. 51. Вагітність 26 тижнів, СЗРП, симетрична форма, ІІІ ст.,
синдром Дауна (абортус)
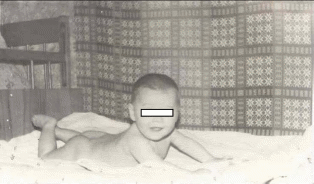
Рис. 52. Синдром Дауна, мозаїчна форма
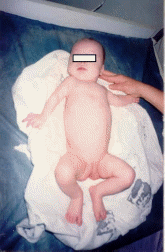
Рис. 53. Синдром Дауна
47,ХХ,+21
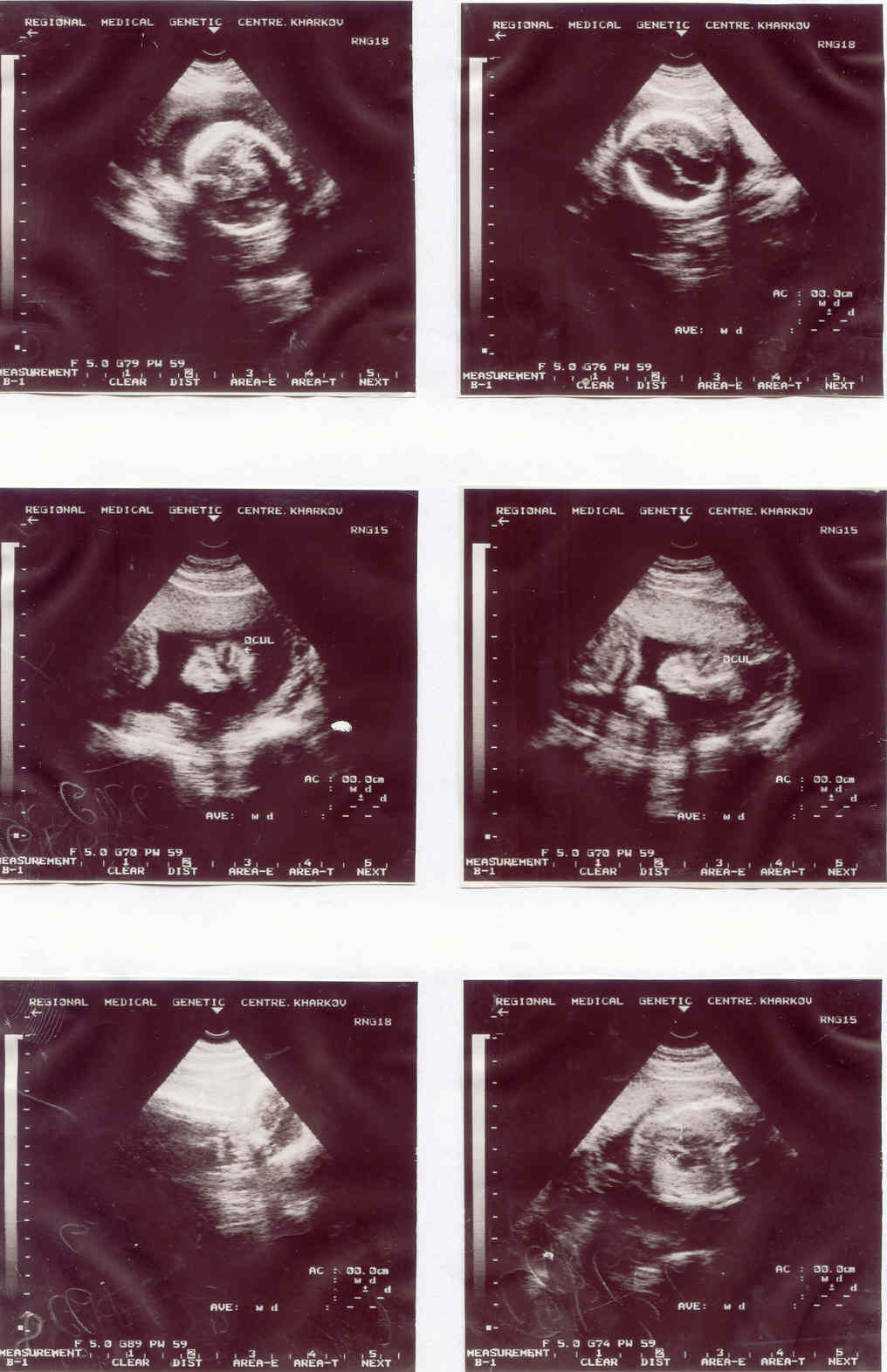
Рис. 54. Синдром Дауна
Ехографічні маркерні ознаки синдрому Дауна (монголоїдний розріз очей, доліхоцефалія, мікроцефалія, ВПС)
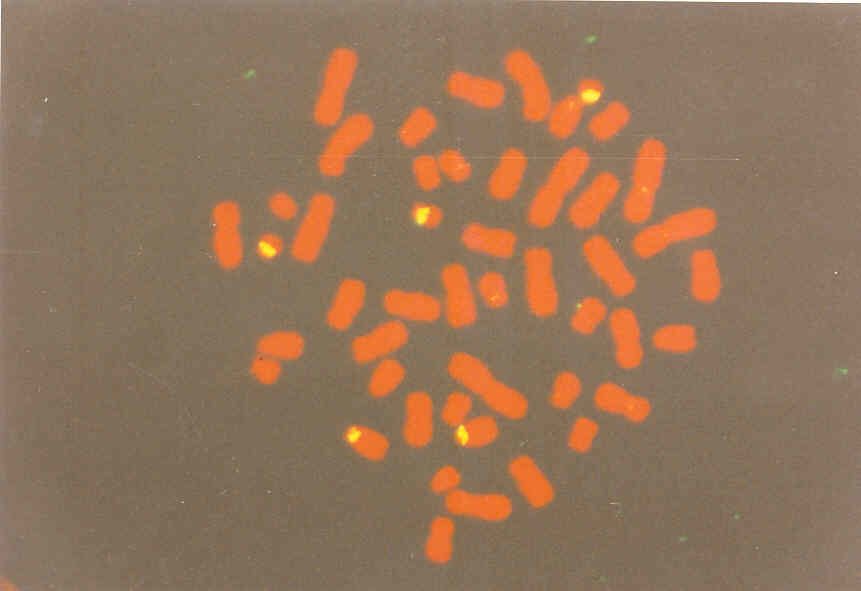
Рис. 55. Пренатальна індентифікація трисомії 21