

Вопрос 6
Периодичность свойств атомов элементов можно проиллюстрировать на самых разных их характеристиках. Перечислим важнейшие из них: радиус атома и атомный объем; потенциал ионизации; сродство к электрону; электроотрицательность атома ; степени окисления; физические свойства соединений (плотность, температуры плавления и кипения).
Радиусы атомов в периоде слева направо уменьшается радиус, а в группах сверху вниз увеличивается. У элементов побочных подгрупп, стоящих после лантанидов и актинидов существенного увеличения атомов не наблюдается в следствии лантаноидного и акиноидного сжатия.
Потенциал (энергия) ионизации I - энергия, необходимая для отрыва наиболее слабо связанного электрона от атома: X → Х+ + е. Наименьшие потенциалы ионизации - у щелочных металлов, наибольшие - у инертных газов.
Сродство к электрону Е - энергия, которая выделяется при присоединении электрона к атому: X + е → X-. Наибольшее сродство к электрону - у галогенов, наименьшее - у металлов. Наибольшим сродством к электрону обладают p-элементы VII группы. Наименьшее сродство к электрону у атомов с конфигурацией s2 (Be, Mg, Zn) и s2p6 (Ne, Ar) или с наполовину заполненными p-орбиталями (N, P, As)
Электроотрицательность –полусумма ионизационного потенциала и сродства к электрону. ЭО- фундаментальное химическое свойство атома, количественная характеристика способности атома в молекуле смещать к себе общие электронные пары. Элeктроотрицательность элементов подчиняется периодическому закону: она растет слева направо в периодах и снизу вверх в главных подгруппах Периодической системы элементов Д.И. Менделеева. Электроотрицательность не является абсолютной константой элемента. Она зависит от эффективного заряда ядра атома, который может изменяться под влиянием соседних атомов или групп атомов, типа атомных орбиталей и характера их гибридизации.
Вертикальная периодичность
Вертикальная периодичность заключается в повторяемости свойств простых веществ и соединений в вертикальных столбцах Периодической системы. Это основной вид периодичности, в соответствии с которым все элементы объединены в группы. Элементы одной группы имеет однотипные электронные конфигурации. Химия элементов и их соединений обычно рассматривается на основе этого вида периодичности. Вертикальная периодичность обнаруживается и в некоторых физических свойствах атомов, например, в энергиях ионизации Ei (кДж/моль)
Горизонтальная периодичность
Горизонтальная периодичность заключается в появлении максимальных и минимальных значений свойств простых веществ и соединений в пределах каждого периода. Она особенно заметна для элементов VIIIБ-группы и лантаноидов (например, лантаноиды с четными порядковыми номерами более распространены, чем с нечетными).В таких физических свойствах, как энергия ионизации и сродство к электрону, также проявляется горизонтальная периодичность, связанная с периодическим изменением числа электронов на последних энергетических подуровнях.
Диагональная периодичность
Диагональная периодичность - повторяемость свойств простых веществ и соединений по диагоналям Периодической системы. Она связана с возрастание неметаллических свойств в периодах слева направо и в группах снизу вверх. Поэтому литий похож на магний, бериллий на алюминий, бор на кремний, углерод на фосфор. Так, литий и магний образуют много алкильных и арильных соединений, которые часто используют в органической химии. Бериллий и алюминий имеют сходные значения окислительно-восстановительных потенциалов. Бор и кремний образуют летучие, весьма реакционноспособные молекулярные гидриды. Диагональную периодичность не следует понимать как абсолютное сходства атомных, молекулярных, термодинамических и других свойств. Та, в своих соединениях атом лития имеет степень окисления (+I), а атом магния - (+II). Однако свойства ионов Li+ и Mg2+ очень близки, проявляясь, в частности, в малой растворимости карбонатов и ортофосфатов.
7) Ковалентная химическая связь. Модель молекулы водорода по Гайтлеру-Лондону. Электронные структуры возбужденных атомов. Спиновая валентность. Метод валентных связей Сущность метода. Основные характеристики ков.св: энергия, длинна, полярность, кратность, насыщаемость. Дипольный момент связи, сигма и пи связи.
К овалентная
связь образуется
за счёт общих электронных пар, возникающих
в оболочках связываемых атомов.
овалентная
связь образуется
за счёт общих электронных пар, возникающих
в оболочках связываемых атомов.
Она может быть образована атомами одного итого же элемента и тогда она неполярная; например, такая ковалентная связь существует в молекулах одноэлементных газов H2, O2, N2, Cl2 и др. |
|
Ковалентная связь может быть образована атомами разных элементов, сходных по химическому характеру, и тогда она полярная; например, такая ковалентная связь существует в молекулах H2O, NF3, CO2. Ковалентная связь образуется между атомами элементов, обладающих электроотрицательным характером.
|
По Гейтлеру-Лондону химическая связь осуществляется в результате взаимодействия спиновых моментов реагирующих атомов. По этой теории для возникновения гомеополярной связи необходимо участие по крайней мере двух электронов, взаимодействующих своими спинами.



Они сближаются и образуют молекулу водорода.

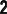
Возбужденные состояния атомов. Образуются из основного состояния при переходе одного или нескольких электронов (напр., под действием излучения) с занятых орбиталей на свободные (или занятые лишь одним электроном).
Возбужденные состояния атома серы. Возбужденные состояния атома фосфора.
Метод валентных связей. В основе ВС метода лежит представление о возникновении связи через образование связующих электронных пар, поэтому в этом методе связь трактуется как двухэлектронная и двуцентровая.
Образование общей пары эл-в может происходить за счет перекрывания одноэлектронных орбиталей взаимодействующих атомов. Этот механизм образования связи наз-ся обменным. С учетом этого механизма валентными электронами в атоме являются непарные электроны, которые могут находиться на внешнем и предвнешнем уровнях.
Второй вариант обр-я общей пары – донорно-акцепторынй механизм. В этом случае перекрываются двухэлектронная орбиталь одного атома и свободная орбиталь второго атома.
Основные характеристики связи в методе ВС следующие: насыщаемость, направленность, кратность, поляризуемость.
Насыщаемость опред-ся числом непарных электронов в атоме и характеризует валентность. Их число может увеличиваться за счет перехода атома в возбужденное состояние.
Направленность связи опред-ся тем, что электронные орбитали атома имеют определенную конфигурацию и ориентацию в пространстве.
Поляризуемость и полярность зависят от различия электроотрицательностей атомов, образующих молекулу. Область перекрывания (общ. электронная пара) смещается в пространстве к более электроотрицательному атому.
Свойства ковалентной связи: насыщаемость, направленность и поляризуемость.
Насыщаемость ковалентной связи обусловлена ограниченными валентными возможностями атомов, т.е. их способностью к образованию строго определенного числа связей, которое обычно лежит в пределах от 1 до 6. Общее число валентных орбиталей в атоме, т.е. тех, которые могут быть использованы для образования химических связей, определяет максимально возможную валентность элемента. Число уже использованных для этого орбиталей определяет валентность элемента в данном соединении.
Н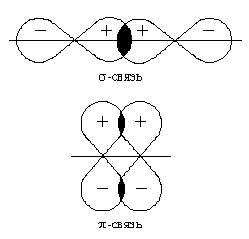 аправленность
ковалентной связи является результатом
стремления атомов к образованию наиболее
прочной связи за счет возможно большей
электронной плотности между ядрами.
Это достигается при такой пространственной
направленности перекрывания электронных
облаков, которая совпадает с их
собственной. Исключение составляют
s-электронные облака, поскольку их
сферическая форма делает все направления
равноценными. Для p- и d-электронных
облаков перекрывание осуществляется
вдоль оси, по которой они вытянуты, а
образующаяся при этом связь называется
s-связью. s-Связь имеет осевую симметрию,
и оба атома могут вращаться вдоль линии
связи, т.е. той воображаемой линии,
которая проходит через ядра химически
связанных атомов.
аправленность
ковалентной связи является результатом
стремления атомов к образованию наиболее
прочной связи за счет возможно большей
электронной плотности между ядрами.
Это достигается при такой пространственной
направленности перекрывания электронных
облаков, которая совпадает с их
собственной. Исключение составляют
s-электронные облака, поскольку их
сферическая форма делает все направления
равноценными. Для p- и d-электронных
облаков перекрывание осуществляется
вдоль оси, по которой они вытянуты, а
образующаяся при этом связь называется
s-связью. s-Связь имеет осевую симметрию,
и оба атома могут вращаться вдоль линии
связи, т.е. той воображаемой линии,
которая проходит через ядра химически
связанных атомов.
Поляризуемость рассматривают на основе представлений о том, что ковалентная связь может быть неполярной (чисто ковалентной) или полярной *.
Важными характеристиками химической связи являются также ее длина и кратность. Длина связи определяется расстоянием между ядрами связанных атомов в молекуле. Как правило, длина химической связи меньше, чем сумма радиусов атомов, за счет перекрывания электронных облаков.
Кратность связи определяется количеством электронных пар, связывающих два атома, например:
этан H3C–CH3 одинарная связь (s-связь)
этилен H2C=CH2 двойная связь (одна s-связь и одна p-связь)
ацетилен HCºCH тройная связь (одна s-связь и две p-связи).
С увеличением кратности возрастает энергия связи, однако это возрастание не пропорционально кратности, т.к. p-связи менее прочны, чем s-связь.
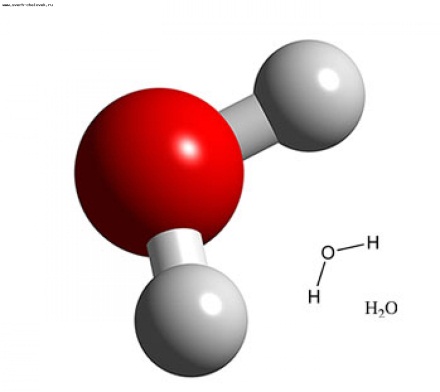
Дипольный момент химической связи обусловлен смещением электронного облака в сторону одного из атомов. Связь называют полярной, если соответствующий дипольный момент существенно отличается от нуля. Возможны случаи, когда отдельные связи в молекуле полярны. а суммарный дипольный момент молекулы равен нулю; такие молекулы наз. неполярными (напр., молекулы СО2 и CCl4). Если же дипольный момент молекулы отличен от нуля, молекула наз. полярной. Напр., молекула Н2О полярна; суммирование дипольных моментов двух полярных связей ОН также дает отличный от нуля дипольный момент, направленный по биссектрисе валентного угла НОН.
Порядок величины дипольный момент молекулы определяется произведением заряда электрона (1,6.10-19 Кл) на длину химической связи (порядка 10-10 м), т. е. составляет 10-29 Кл.м. В справочной литературе дипольный момент молекул приводят в дебаях (Д или D), по имени П. Дебая; 1 Д = 3,33564.10-30 Кл.м.
8) Обменный и донорно-акцепторный механизмы образования связи. Перекрывания атомных орбиталей связующих атомов. Донор и акцептор. Хим. св. в частицах HF, Cl2, NH4+. Валентные возможности атомов элементов II и III периодов. Максимальная валентность.
В методе валентных связей различают обменный и донорно-акцепторный механизмы образования химической связи.
Обменный механизм. К обменному механизму образования химической связи относятся случаи, когда в образовании электронной пары от каждого атома участвует по одному электрону.
В молекулах Н2, Li2, Na2 связи образуются за счет неспаренных s-электронов атомов. В молекулах F2 и Cl2 – за счет неспаренных р-электронов. В молекулах HF и HCl связи образуются s-электронами водорода и p-электронами галогенов.
Особенностью образования соединений по обменному механизму является насыщаемость, которая показывает, что атом образует не любое, а ограниченное количество связей. Их число, в частности, зависит от количества неспаренных валентных электронов.





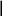

Донорно-акцепторный механизм образования связи – механизм образования связи, при котором один из связываемых атомов является донором электронной пары, а другой – акцептором.
Атом, который представляет свободную орбиталь, называется акцептором, атом, представляющий электронную пару, является донором.
Условия образования связи по донорно-акцепторному механизму:
1) наличие у одного из атомов неподеленной пары валентных электронов;
2) наличие у другого атома свободной орбитали на валентном подуровне.
Донорно-акцепторный механизм образования связи распространен довольно широко. Особенно часто он встречается при образовании соединений d-элементов. Атомы почти всех d-элементов имеют много свободных валентных орбиталей. Поэтому они являются активными акцепторами электронных пар.
Известно, что валентно насыщенные соединения аммиак NH3 и трифторид бора BF3 реагируют друг с другом по реакции
NH3 + BF3 = NH3BF3 + 171,4 кДж/моль.
Р ассмотрим
механизм этой реакции:
ассмотрим
механизм этой реакции:
Видно, что из четырех орбиталей бора три заселены, а одна - остается вакантной. В молекуле аммиака заселены все четыре орбитали азота, из них три – по обменному механизму электронами азота и водорода, а одна содержит электронную пару, оба электрона которой принадлежат азоту. Такая электронная пара называется неподеленной электронной парой. Образование соединения H3N · BF3 происходит за счет того, что неподеленная электронная пара аммиака занимает вакантную орбиталь фторида бора. При этом уменьшается потенциальная энергия системы и выделяется эквивалентное количество энергии. Подобный механизм образования называют донорно-акцепторным, донором – такой атом, который отдает свою электронную пару для образования связи (в данном случае атом азота); а атом, который предоставляя вакантную орбиталь, принимает электронную пару, называется акцептором (в данном случае атом бора). Донорно-акцепторная связь является разновидностью ковалентной связи.
В соединении H3N · BF3 азот и бор – четырехвалентны. Атом азота повышает свою валентность от 3 до 4 в результате использования неподеленной электронной пары для образования дополнительной химической связи. Атом бора повышает валентность за счет наличия у него свободной орбитали на валентном электронном уровне. Таким образом, валентность элементов определяется не только числом неспаренных электронов, но и наличием неподеленных электронных пар и свободных орбиталей на валентном электронном уровне.
Более простым случаем образования химической связи по донорно-акцепторному механизму является реакция аммиака с ионом водорода:
Роль акцептора электронной пары играет пустая орбиталь иона водорода. В ионе аммония NH4+ атом азота четырехвалентен.
Перекрывание атомных орбиталей
При описании электронного строения химической частицы электроны, в том числе и обобществленные, относят к отдельным атомам и их состояния описывают атомными орбиталями. При решении уравнения Шредингера приближенную волновую функцию выбирают так, чтобы она давала минимальную электронную энергию системы, то есть наибольшее значение энергии связи. Это условие достигается при наибольшем перекрывании орбиталей, принадлежащей одной связи. Таким образом, пара электронов, связывающих два атома, находится в области перекрывания их атомных орбиталей.
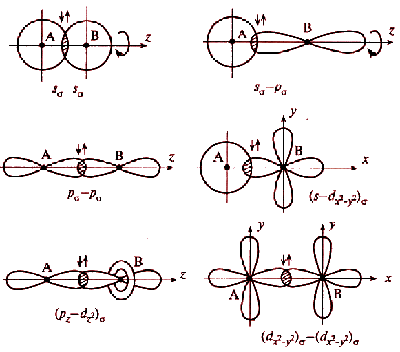
Перекрываемые орбитали должны иметь одинаковую симметрию относительно межъядерной оси.
Перекрывание атомных орбиталей вдоль линии, связывающей ядра атомов, приводит к образованию σ-связей. Между двумя атомами в химической частице возможна только одна σ-связь. Все σ-связи обладают осевой симметрией относительно межъядерной оси. Фрагменты химических частиц могут вращаться вокруг межъядерной оси без нарушения степени перекрывания атомных орбиталей, образующих σ-связи. Совокупность направленных, строго ориентированных в пространстве σ-связей создает структуру химической частицы.
При дополнительном перекрывании атомных орбиталей, перпендикулярных линии связи, образуются π-связи.
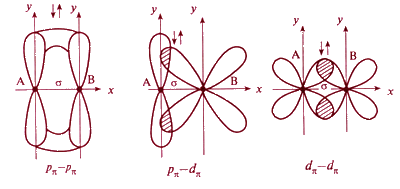
В результате этого между атомами возникают кратные связи:
Одинарная (σ) |
Двойная (σ +π) |
Тройная (σ + π + π) |
F−F |
O=O |
N≡N |
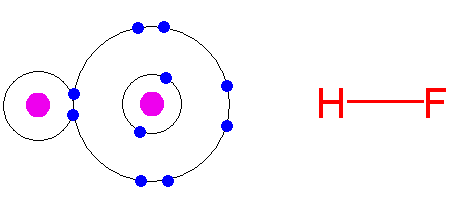
Химическая связь в частицах:
H
 F
- между атомами водорода и фтора
образуется ковалентная полярная
химическая связь. Из-за того что фтор
более электроотрицателен чем водород,
фтор притянет к себе электронную пару.
F
- между атомами водорода и фтора
образуется ковалентная полярная
химическая связь. Из-за того что фтор
более электроотрицателен чем водород,
фтор притянет к себе электронную пару.
Во фтороводороде образуется водородная связь: HF---HF, где донором электронов является атом фтора. Процесс образования водородной связи при взаимодействии двух молекул HF можно представить следующей схемой:
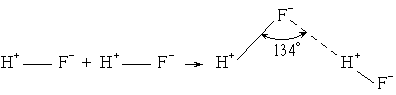
Cl2 - два атома хлора в молекуле Cl2 образуют ковалентную неполярную связь по обменному механизму, объединяя свои неспаренные 3р-электроны.
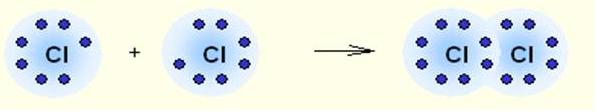
NH4+ - заряженная частица. Образование связи у нее проходит по донорно-акцепторному механизму. Связь ковалентная полярная.
Валентные возможности атомов элементов II и III периодов. Максимальная валентность.
Строение наружных энергетических уровней атомов химических элементов и определяет в основном свойства их атомов. Поэтому эти уровни называют валентными. Электроны этих уровней, а иногда и предвнешних уровней могут принимать участие в образовании химических связей. Такие электроны также называют валентными.
Валентность атома химического элемента определяется в первую очередь числом неспаренных электронов, принимающих участие в образовании химической связи.
Валентные электроны атомов элементов главных подгрупп расположены на s- и р-орбиталях внешнего электронного слоя. У элементов побочных подгрупп, кроме лантаноидов и актиноидов, валентные электроны расположены на s-орбитали внешнего и d-орбиталях предвнешнего слоев.
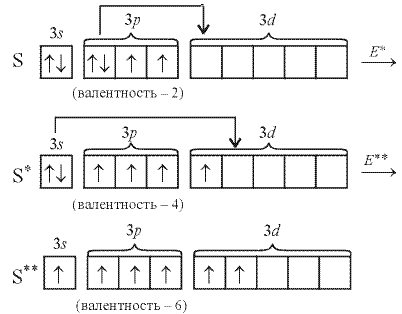
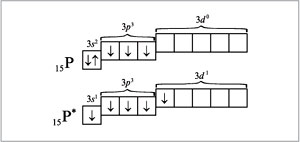
Для того чтобы верно оценить валентные возможности атомов химических элементов, нужно рассмотреть распределение электронов в них по энергетическим уровням и подуровням и определить число неспаренных электронов в соответствии с принципом Паули и правилом Хунда для невозбужденного (основного, или стационарного) состояния атома и для возбужденного (то есть получившего дополнительную энергию, в результате чего происходит распаривание электронов внешнего слоя и переход их на свободные орбитали). Атом в возбужденном состоянии обозначают соответствующим символом элемента со звездочкой. Например, рассмотрим валентные возможности атомов фосфора в стационарном и возбужденном состояниях:
![]()
В невозбужденном состоянии атом фосфора имеет три не-спаренных электрона на р-подуровне. При переходе атома в возбужденное состояние один из пары электронов s-подуровня может переходить на свободную орбиталь d-подуровня. Валентность фосфора при этом изменяется с трех (в основном состоянии) до пяти (в возбужденном состоянии).



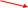
Так, атом углерода в стационарном состоянии имеет два неспаренных электрона. Следовательно, с их участием могут образоваться две общие электронные пары, осуществляющие две ковалентные связи. Однако вам хорошо известно, что во многих неорганических и во всех органических соединениях присутствуют атомы четырехвалентного углерода. Очевидно, что его атомы образовали четыре ковалентные связи в этих соединениях, находясь в возбужденном состоянии.
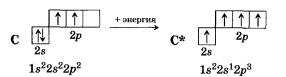
Валентные возможности атомов химических элементов далеко не исчерпываются числом неспаренных электронов в стационарном и возбужденном состояниях атомов. Если вы вспомните донорно-ак-цепторный механизм образования ковалентных связей, то вам станут понятны и две другие валентные возможности атомов химических элементов, которые определяются наличием свободных орбиталей и наличием неподеленных электронных пар, способных дать ковалентную химическую связь по донор-но-акцепторному механизму.
Вывод:
Валентные возможности атомов химических элементов определяются:
1) числом неспаренных электронов (одноэлектронных орбиталей);
2) наличием свободных орбиталей;
3) наличием неподеленных пар электронов.
Максимальную валентность элемента считают равной числу электронов во внеш. Электронной оболочке атома. Максимальная валентность элементов одной и той же группы периодич. Системы обычно соответствует ее порядковому номеру.
9. Направленность химической связи. Гибридизация атомных орбиталей. Типы гибридизации. Геометрическая форма и полярность молекул. Нелокализованная химическая связь. Тип гибридизации и геометрия частиц BeF2, СO2, CO32-, С2H4, H2O, NH3, SO42-.
Направленность связи - обусловлена пространственным расположением атомов в многоатомных частицах (радикалах, молекулах, ионах) и немолекулярных химических соединениях (например, кристаллах). Обычно ее связывают с гибридизацией атомных орбиталей. Направленность ковалентных связей обуславливает явление изомерии и определяет [кристалло]химическое строение вещества. Если перекрывание s-облаков происходит вдоль линии, соединяющей взаимодействующие ядра атомов, возникающая ковалентная связь называется σ-связью. Например, при образовании молекулы фтороводорода (HF) происходит перекрывание s-облака атома водорода (Н) с р-облаком атома фтора (F). При образовании π-связи осуществляется так называемое боковое перекрывание электронных облаков, и плотность электронного облака максимальна «над» и «под» плоскостью σ-связи.
Теория валентных связей (ВС) предполагает участие в образовании ковалентных связей не только "чистых" АО, но и "смешанных", так называемых гибридных, АО. При гибридизации первоначальная форма и энергия орбиталей (электронных облаков) взаимно изменяются и образуются орбитали (облака) новой одинаковой формы и одинаковой энергии. Число гибридных орбиталей равно числу исходных.
Г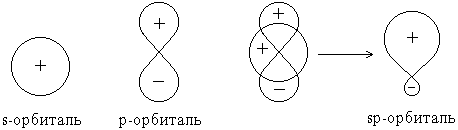 ибридная
орбиталь, возникшая за счет комбинации
s-
и p-орбиталей.
Она больше вытянута по одну сторону от
ядра, чем по другую, т.е. электронная
плотность в ней сконцентрирована по
одну сторону в большей степени, чем по
другую.
ибридная
орбиталь, возникшая за счет комбинации
s-
и p-орбиталей.
Она больше вытянута по одну сторону от
ядра, чем по другую, т.е. электронная
плотность в ней сконцентрирована по
одну сторону в большей степени, чем по
другую.
s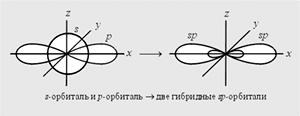 p
p
1 80°
80°
линейная
H–Be–H, HC≡CH
sp2
1 20°
20°
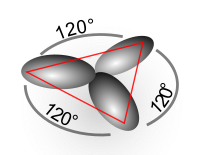
плоская тригональная
H2C=CH2, C6H6, BCl3
s p3
p3
1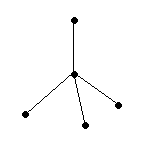 09°28'
09°28'
тетраэдрическая
[NH4]+, CH4, CCl4, H3C–CH3
sp2d
9 0°
0°
квадратная
[Ni(CN)4]2–, [PtCl4]2–
sp3d или dsp3
9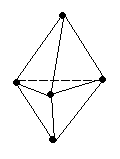 0°,
120°
0°,
120°
триагонально-бипирамидальная
PCl5
d2sp3 или sp3d2
9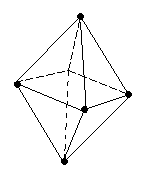 0°
0°
октаэдрическая
[Fe(CN)6]3–, [CoF6]3–, SF
Гибридизация и геометрия частиц:
BeF2
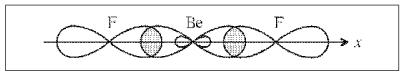
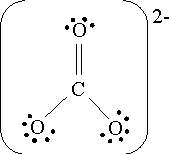
СО2
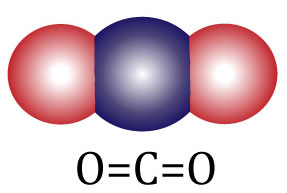
С
 О3
О3
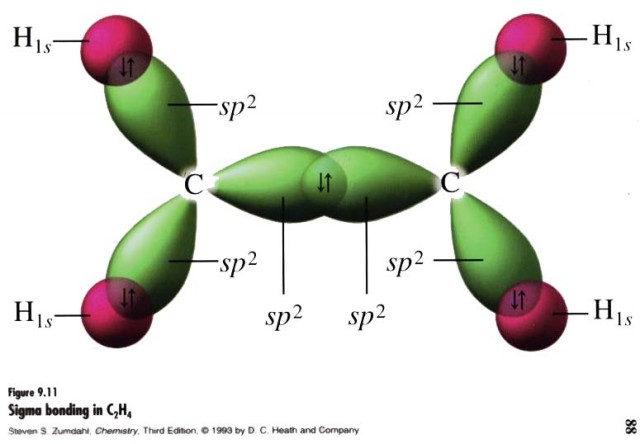
С2Н4
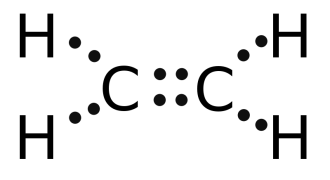
Н2О
Билет 10.Ионная химическая связь, её основные характеристики. Примеры молекул. Ионные Кристаллические решетки. Координационное число. Поляризация ионов. Поляризующее действие и поляризуемость. Влияние поляризации на свойства веществ.
Ионная связь является разновидностью ковалентной сильно полярной связи. Ионная связь — химическая связь, образующаяся между атомами с большой разностью электроотрицательностей, при которой общая электронная пара полностью переходит к атому с большей электроотрицательностью. Ионная связь характерна для соединений типичных металлов(щелочные, щелочноземельные металлы) с типичными неметаллами(галогены,кислород).Ионная связь является достаточно прочной. Основные характеристики ионной химической связи:1. Ненаправленность 2. ненасыщаемость. Примеры молекул:NaCl, CsF.
Ионные кристаллические решетки: В узлах кристаллической решетки находятся противоположно заряженные ионы- катионы и анионы. Для веществ с ионной решеткой характерны достаточно высокие температуры плавления, под действием растворителей происходит разрушение решетки.
Координационное число.Вследствие ненаправленности и ненасыщаемости ионной связи энергетически наиболее выгодно, когда каждый ион окружен максимальным числом ионов противоположного знака. Однако из-за отталкивания одноименных ионов друг от друга устойчивость системы достигается лишь при определенной взаимной координации ионов.Координационное число зависит от соотношения размеров ионов.
Поляризация ионов выражается в относительном смещении ядра и окружающих его электронов внешней электронной оболочки под действием электрического поляч соседнего иона, при этом валентные электроны смещаются в строну катионов.
Поляризующее действие иона т.е его способность деформировать, поляризовать другой ион и сильно зависит от его электронной структуры.Ионы с 8 ми электронной структурой атомов элементов инертных газов оказывают более слабое поляризующее действие, чем ионы с незавершенным электронным слоем.Наиболее сильное поляризующее действие проявляют ионы с 18-электронной структурой внешнего слоя(Cu+ , Ag+, Zn+)
Поляризуемость ионов, т.е их способность деформироваться под действие внешнего электрического поля,Ю характеризуется следующими особенностями:1.При одинаково абсолютном значении заряда и равных радиусах ионов поляризуемость анионов больше поляризуемости катионов.2.Поляризуемость ионов со сходственным электронным строение возрастает с ростом ионного радиуса, т.е с увеличение числа электронных слоев.3.При одном и том же заряде и одинаковом радиусе ионов поляризуемость ионов 18-электронной оболочкой (Cu+)выше,чем с 8-электронной структурой атомов элементов инертных газов(Na+, Ca+)
Чем более поляризована частица, тем ниже энергия электронных переходов. Если поляризация мала, возбуждение электронов требует более высокой энергии, что отвечает ультрафиолетовой части спектра. Такие вещества обычно бесцветны. В случае сильной поляризации ионов возбуждение электронов происходит при поглощении электромагнитного излучения видимой области спектра. Поэтому некоторые вещества, образованные бесцветными ионами, окрашены.
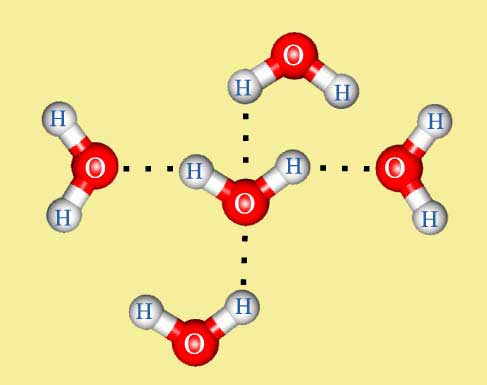
Билет 11.Водородная связь, образование, влияние на свойства веществ.Водородная связь – это связь между положительно заряженным атомом водорода одной молекулы и отрицательно заряженным атомом другой молекулы. Водородная связь имеет частично электростатический, частично донорно- акцепторный характер. Водородная связь характерна для соединений водорода с наиболее ЭО элементами: фтора, кислорода, азоты и в меньшей степени хлора и серы. Образование водородной связи обязано ничтожно малому размеру положительно поляризованного атома водорода и его способности глубоко внедряться в электронную оболочку соседнего(ковалентно с ним не связанного) отрицательно поляризованного атома. Наличие водородных связей объсняет высокие температуры кипения воды, спиртов, карбоновых кислот. Образование межмолекулярных водородных связей приводит к существенному изменению свойств веществ: повышению вязкости, температур плавления и кипения, теплот парообразования и плавления. Например, вода, фтороводород и аммиак имеют аномально высокие температуры кипения и плавления. Под влиянием водородных связей изменяются и химические свойства.
Билет 12.Межмолекулярное взаимодействие: ориентационное, поляризационное и дисперсионное.
Ван-дер-ваальсовы силы — силы межмолекулярного взаимодействия с энергией 0,8 — 8,16 кДж/моль. Этим термином первоначально обозначались все такие силы, в современной науке он обычно применяется к силам, возникающим при поляризации молекул и образовании диполей. Открыты Я. Д. ван дер Ваальсом в 1869 году.
Ван-дер-ваальсовое взаимодействие состоит из трех типов слабых взаимодействий:
Ориентационные силы, диполь-дипольное притяжение. Осуществляется между молекулами, являющимися постоянными диполями. Примером может служить HCl в жидком и твердом состоянии. Энергия такого взаимодействия обратно пропорциональна кубу расстояния между диполями.
Дисперсионное притяжение (лондоновские силы). Взаимодействием между мгновенным и наведенным диполем. Энергия такого взаимодействия обратно пропорциональна шестой степени расстояния между диполями.
Индукционное притяжение. Взаимодействие между постоянным диполем и наведенным (индуцированным). Энергия такого взаимодействия обратно пропорциональна шестой степени расстояния между диполями.
Билет 13
Химическая термодинамика
Хими́ческая термодина́мика — раздел физической химии, изучающий процессы взаимодействия веществ , а также зависимости термодинамических свойств веществ от их состава и агрегатного состояния..
Основными направлениями химической термодинамики являются:
1. Классическая химическая термодинамика, изучающая термодинамическое равновесие вообще.
2. Термохимия, изучающая тепловые эффекты, сопровождающие химические реакции.
3. Теория растворов, моделирующую термодинамические свойства вещества исходя из представлений о молекулярном строении и данных о межмолекулярном взаимодействии.
Химическая термодинамика определяет, в первую очередь, условия (такие, как температура и давление) протекания химических реакций и равновесных состояний, которых они достигают. Анализ тепловых явлений базируется на трех фундаментальных принципах, подтвержденных данными многочисленных наблюдений.
Системы и их классификация
Система – тело или несколько тел, находящихся во взаимодействии между собой (диффузия, теплообмен, химическая реакция) и отделенных от окружающей среды.
Состояние системы в термодинамике определяется с помощью набора переменных, называемых параметрами состояния и характеризующих термодинамическое состояние при равновесии. Всякое изменение, происходящее в системе и связанное с изменением хотя бы одного из параметров состояния, называется термодинамическим процессом.
Системы имеют определенные границы, отделяющие их от внешней среды, и могут быть гомогенными или гетерогенными.
Гомогенная система – система, в которой все макроскопические свойства в любых ее частях имеют одно и то же значение или непрерывно меняются от точки к точке. Примеры: ненасыщенные растворы, пар, газовые смеси. Составленные части гомогенной системы не могут быть выделены из нее с помощью простых механических приемов (фильтрования, отбора и т. д.).
Гетерогенная система – система, составные части которой отделены друг от друга видимыми поверхностями раздела, на которых происходят резкие скачкообразные изменения какого-либо свойства. Примеры: насыщенный раствор какой-либо соли, находящийся в равновесии с кристаллами этой соли, две несмешивающиеся жидкости и т. д.). Составные части таких систем могут быть отделены друг от друга с помощью механических операций.
Совокупность тел, энергетически взаимодействующих между собой и с другими телами, обменивающихся с ними веществом, называется термодинамической системой.
Системы делят на изолированные (это те системы, которые не обмениваются энергией и веществом с другими системами), открытые (те системы, которые обмениваются с окружающей средой и веществом, и энергией), закрытые (системы, в которых есть только обмен энергией).
Термодинамические параметры и ф-я состояния системы.
Любая ТДС характеризуется параметрами: температура, давление, плотность, концентрация, мольный объем. В любой ТДС обязательно протекают процессы, и они могут быть равновесными, неравновесными, обратимыми и необратимыми.
Если в ТДС определенное свойство системы не будет изменяться во времени, т. е. оно будет одинаковым во всех точках объема, то такие процессы – равновесные.
В неравновесных процессах свойство системы будет изменяться во времени без воздействия окружающей среды.
Обратимые процессы – процессы, в которых система возвращается в первоначальное состояние.
Необратимые – когда система не возвращается в первоначальное состояние.
Функции могут зависеть от пути процесса. Функции, которые зависят от начального и конечного состояний системы и не зависят от пути процесса, – функции состояния; внутренняя энергия, энтальпия, энтропия и другие – полные дифференциалы.
Первое начало термодинамики
Первое начало термодинамики, один из двух основных законов термодинамики, представляет собой закон сохранения энергии для систем, в которых существенное значение имеют тепловые процессы. Согласно П. н. т., термодинамическая система (например, пар в тепловой машине) может совершать работу только за счёт своей внутренней энергии или каких-либо внешних источников энергии. П. н. т. часто формулируют как невозможность существования вечного двигателя 1-го рода, который совершал бы работу, не черпая энергию из какого-либо источника.
При сообщении термодинамической системе некоторого количества теплоты Q в общем случае происходит изменение внутренней энергии системы DU и система совершает работу А:
Q = DU + A (1)
Уравнение (1), выражающее П. н. т., является определением изменения внутренней энергии системы (DU), так как Q и А — независимо измеряемые величины.
Внутреннюю энергию системы U можно, в частности, найти, измеряя работу системы в адиабатном процессе (то есть при Q = 0): Аад = — DU, что определяет U с точностью до некоторой аддитивной постоянной U0:
U = U + U0 (2)
П. н. т. утверждает, что U является функцией состояния системы, то есть каждое состояние термодинамической системы характеризуется определённым значением U, независимо от того, каким путём система приведена в данное состояние (в то время как значения Q и А зависят от процесса, приведшего к изменению состояния системы).
Вну́тренняя эне́ргия.
Вну́тренняя эне́ргия тела (обозначается как E или U) — это сумма энергий молекулярных взаимодействий и тепловых движений молекулы. Внутренняя энергия является однозначной функцией состояния системы. Это означает, что всякий раз, когда система оказывается в данном состоянии, её внутренняя энергия принимает присущее этому состоянию значение, независимо от предыстории системы. Следовательно, изменение внутренней энергии при переходе из одного состояния в другое будет всегда равно разности между ее значениями в конечном и начальном состояниях, независимо от пути, по которому совершался переход.
Билет14
Термохимия.
Термохимия - раздел химической термодинамики, в задачу которой входит определение и изучение тепловых эффектов реакций, а также установление их взаимосвязей с различными физико-химическими параметрами. Ещё одной из задач термохимии является измерение теплоёмкостей веществ и установление их теплот фазовых переходов.
Почти каждая химическая реакция связана с тем или иным тепловым эффектом: химическое превращение сопровождается или выделением, или поглощением тепла. В первом случае реакция называется экзотермической, во втором - эндотермической. Тепловой эффект реакций является мерилом изменения внутренней энергии тел, участвующих в химическом превращении. Взаимные превращения различных видов энергии составляют предмет термодинамики, установившей законы таких превращений; поэтому в основе Т., занимающейся одним из видов таких превращений, лежат законы термодинамики, приложение которых к химическим явлениям и составляет содержание Т. Соответственно этому, дальнейшее изложение разделено на три части: очерк развития Т., приложения I закона и приложения II закона термодинамики.
Термохимические уравнения.
Важнейшей величиной в термохимии является стандартная теплота образования (стандартная энтальпия образования). Стандартной теплотой (энтальпией) образования сложного вещества называется тепловой эффект (изменение стандартной энтальпии) реакции образования одного моля этого вещества из простых веществ в стандартном состоянии. Стандартная энтальпия образования простых веществ в этом случае принята равной нулю.
В термохимических уравнениях необходимо указывать агрегатные состояния веществ с помощью буквенных индексов, а тепловой эффект реакции (ΔН) записывать отдельно, через запятую.
В термохимии также используют уравнения, в которых тепловой эффект относят к одному молю образовавшегося вещества, применяя в случае необходимости дробные коэффициенты.
Стандартное состояние в термохимии - состояние вещества, в котором оно находится при температуре 298,15 К и давлении 101,325 кПа
Тепловой эффект реакции.
Энергетический эффект хим. процесса возникает при изменении в системе внутренней энергии или энтальпии. Для хим. реакций под работой против внешних сил подразумевается работа против внешнего давления. Обычно она совершенствуется за счёт расширения системы. Работа против внешнего давления равна произведению давления на изменение объёма системы, при переходе из одного состояния в другое. Если хим. реакция протекает при постоянном объёме, то выделение или поглощение теплоты связано с изменением внутренней энергии системы.
Энергетический эффект реакции, протекающий при пост давлении, отличается от энергетического эффекта реакции, протекающим при постоянном объёме. Подавляющее большинство реакций протекают при пост давлении.
Закон Гесса
Закон Гесса — основной закон термохимии, который формулируется следующим образом:
• Тепловой эффект химической реакции, проводимой в изобарно-изотермических или изохорно-изотермических условиях, зависит только от вида и состояния исходных веществ и продуктов реакции и не зависит от пути её протекания.
Иными словами, количество теплоты, выделяющееся или поглощающееся при каком-либо процессе, всегда одно и то же, независимо от того, протекает ли данное химическое превращение в одну или в несколько стадий (при условии, что температура, давление и агрегатные состояния веществ одинаковы). Например, окисление глюкозы в организме осуществляется по очень сложному многостадийному механизму, однако суммарный тепловой эффект всех стадий данного процесса равен теплоте сгорания глюкозы.
На рисунке приведено схематическое изображение некоторого обобщенного химического процесса превращения исходных веществ А1, А2… в продукты реакции В1, В2…, который может быть осуществлен различными путями в одну, две или три стадии, каждая из которых сопровождается тепловым эффектом ΔHi. Согласно закону Гесса, тепловые эффекты всех этих реакций связаны следующим соотношением:
ΔH1 = ΔH2 + ΔH3 = ΔH4 + ΔH5 + ΔH6
Закон открыт русским химиком Г. И. Гессом в 1840 г.; он является частным случаем первого начала термодинамики применительно к химическим реакциям. Практическое значение закона Гесса состоит в том, что он позволяет рассчитывать тепловые эффекты самых разнообразных химических процессов; для этого обычно используют ряд следствий из него.
Следствия из закона Гесса
Тепловой эффект прямой реакции равен по величине и противоположен по знаку тепловому эффекту обратной реакции (закон Лавуазье — Лапласа).
Тепловой эффект химической реакции равен разности сумм теплот образования (ΔHf) продуктов реакции и исходных веществ, умноженных на стехиометрические коэффициенты (ν):
Тепловой эффект химической реакции равен разности сумм теплот сгорания (ΔHc) исходных веществ и продуктов реакции, умноженных на стехиометрические коэффициенты (ν):
Таким образом, пользуясь табличными значениями теплот образования или сгорания веществ, можно рассчитать теплоту реакции, не прибегая к эксперименту. Табличные величины теплот образования и сгорания веществ обычно относятся к т. н. стандартным условиям. Для расчёта теплоты процесса, протекающего при иных условиях, необходимо использовать и другие законы термохимии, например, закон Кирхгофа, описывающий зависимость теплового эффекта реакции от температуры.
Если начальное и конечное состояния химической реакции (реакций) совпадают, то ее (их) тепловой эффект равен нулю.
Стандартная энтальпия в-ва и энтальпия реакции.
Под стандартной теплотой образования понимают тепловой эффект реакции образования одного моля вещества из простых веществ, его составляющих, находящихся в устойчивых стандартных состояниях.
Энтальпия образования простых веществ принимается равной нулю, причем нулевое значение энтальпии образования относится к агрегатному состоянию, устойчивому при T = 298 K.
энтальпия реакции ΔH - это тепловой эффект реакции при постоянном давлении
ΔH = ΔU + pΔV
Единица энтальпии в СИ джоуль (Дж); в химии и справочных таблицах чаще используется кратная единица - килоджоуль (кДж).
Встречающееся в старой литературе обозначение теплового эффекта реакции через Q (без индекса) обычно относится к условию p = const, т.е. характеризует энтальпию реакции ΔH.
15 Билет
Второе начало термодинамики — физический принцип, накладывающий ограничение на направление процессов передачи тепла между телами.Второе начало термодинамики гласит, что невозможен самопроизвольный переход тепла от тела, менее нагретого, к телу, более нагретому.Второе начало термодинамики запрещает так называемые вечные двигатели второго рода, показывая что коэффициент полезного действияне может равняться единице, поскольку для кругового процесса температура холодильника не должна равняться 0.Второе начало термодинамики является постулатом, не доказываемым в рамках термодинамики. Оно было создано на основе обобщения опытных фактов и получило многочисленные экспериментальные подтверждения.
Термодинамическая вероятность — число способов, которыми может быть реализовано состояние физической системы. В термодинамике состояние физической системы характеризуется определёнными значениями плотности, давления, температуры и др. измеримых величин. Перечисленные величины определяют состояние системы в целом (её макросостояние). Однако при одной и той же плотности, температуре и т. д. частицы системы могут различными способами распределиться в пространстве и иметь различные импульсы. Каждое данное распределение частиц называется микросостоянием системы. Вероятность термодинамическая (обозначается W) равна числу микросостояний, реализующих данное макросостояние, из чего следует, что W3 = 1 . Вероятность термодинамическая связана с одной из основных макроскопических характеристик системыэнтропией S соотношением Больцмана: S = k * ln(W), где k — Больцмана постоянная.
Энтропи́я (от греч. ἐντροπία — поворот, превращение) в естественных науках — мера беспорядка системы, состоящей из многихэлементов. В частности, в статистической физике — мера вероятности осуществления какого-либо макроскопического состояния; в теории информации — мера неопределённости какого-либо опыта (испытания), который может иметь разные исходы, а значит и количествоинформации; в исторической науке, для экспликации феномена альтернативности истории (инвариантности и вариативности исторического процесса).Понятие энтропии впервые было введено Клаузиусом в термодинамике в 1865 году для определения меры необратимого рассеивания энергии, меры отклонения реального процесса от идеального. Определённая как сумма приведённых теплот, она является функцией состояния и остаётся постоянной при обратимых процессах, тогда как в необратимых — её изменение всегда положительно.
г![]() де dS —
приращение энтропии; δQ —
де dS —
приращение энтропии; δQ —
минимальная теплота подведенная к
системе; T — абсолютная температура процесса;
Энтропия,
функция состояния S термодинамической
системы, изменение которой dS для
бесконечно малого обратимого изменения
состояния системы равно отношению
количества теплоты ![]() полученного
системой в этом процессе (или отнятого
от системы), к абсолютной температуре Т:
полученного
системой в этом процессе (или отнятого
от системы), к абсолютной температуре Т:
![]()
Величина dS является полным дифференциалом, т.е. ее интегрирование по любому произвольно выбранному пути дает разность между значениями энтропии в начальном (А) и конечном (В) состояниях:
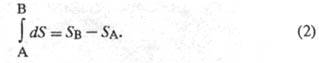
Теплота не является функцией состояния, поэтому интеграл от δQ зависит от выбранного пути перехода между состояниями А и В. Энтропия измеряется в Дж/(моль·град).
Свободная энергия Гиббса (или просто энергия Гиббса, или потенциал Гиббса, или термодинамический потенциал в узком смысле) — это величина, показывающая изменение энергии в ходе химической реакции и дающая таким образом ответ на принципиальную возможность химической реакции; это термодинамический потенциал следующего вида:
![]()
Энергию Гиббса можно понимать как полную химическую энергию системы (кристалла, жидкости и т. д.)
Понятие энергии Гиббса широко используется в термодинамике и химии.