

[Править] Водородная связь в нуклеиновых кислотах и белках
Водородная связь в значительной мере определяет свойства и таких биологически важных веществ, как белки и нуклеиновые кислоты. В частности, элементы вторичной структуры (например, α-спирали, β-складки) и третичной структуры в молекулах белков, РНК и ДНК стабилизированы водородными связями. В этих макромолекулах, водородные связи сцепляют части той же самой макромолекулы, заставляя её сворачиваться в определенную форму. Например, двойная спиральная структура ДНК, определяется в значительной степени наличием водородных связей, сцепляющих пары нуклеотидов, которые связывают одну комплементарную нить с другой.
[Править] Водородная связь в полимерах
Много полимеров усилены водородными связями в их главных цепях. Среди синтетических полимеров самый известный пример - нейлон, где водородные связи играют главную роль в кристаллизации материала. Водородные связи также важны в структуре полученных искусственно полимеров (например, целлюлозы) и в многих различных формах в природе, таких как древесина, хлопок и лён.
Структура воды
Молекула воды представляет собой маленький диполь, содержащий положительный и отрицательный заряды на полюсах. Так как масса и заряд ядра кислорода больше чем у ядер водорода, то электронное облако стягивается в сторону кислородного ядра. При этом ядра водорода "оголяются". Таким образом, электронное облако имеет неоднородную плотность. Около ядер водорода имеется недостаток электронной плотности, а на противоположной стороне молекулы, около ядра кислорода, наблюдается избыток электронной плотности. Именно такая структура и определяет полярность молекулы воды. Если соединить прямыми линиями эпицентры положительных и отрицательных зарядов получится объемная геометрическая фигура - правильный тетраэдр.
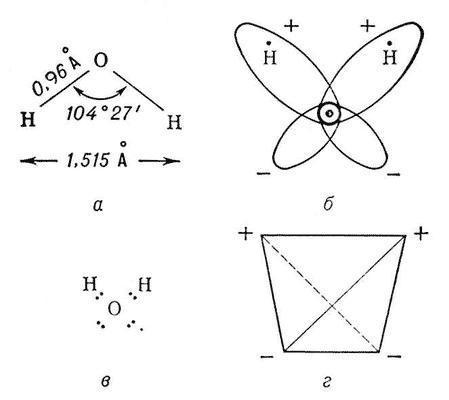
Благодаря наличию водородных связей каждая молекула воды образует водородную связь с 4-мя соседними молекулами, образуя ажурный сетчатый каркас в молекуле льда. Однако, в жидком состоянии вода - неупорядоченная жидкость; эти водородные связи - спонтанные, короткоживущие, быстро рвутся и образуются вновь. Всё это приводит к неоднородности в структуре воды.
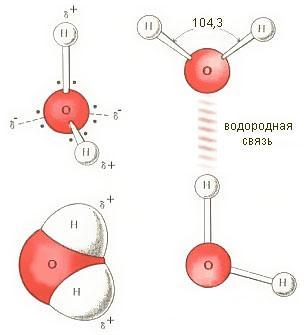
То, что вода неоднородна по своему составу, было установлено давно. С давних пор известно, что лёд плавает на поверхности воды, то есть плотность кристаллического льда меньше, чем плотность жидкости.
Почти у всех остальных веществ кристалл плотнее жидкой фазы. К тому же и после плавления при повышении температуры плотность воды продолжает увеличиваться и достигает максимума при 4°C. Менее известна аномалия сжимаемости воды: при нагреве от точки плавления вплоть до 40°C она уменьшается, а потом увеличивается. Теплоёмкость воды тоже зависит от температуры немонотонно.
Кроме того, при температуре ниже 30°C с увеличением давления от атмосферного до 0,2 ГПа вязкость воды уменьшается, а коэффициент самодиффузии - параметр, который определяет скорость перемещения молекул воды относительно друг друга растёт.
Для других жидкостей зависимость обратная, и почти нигде не бывает, чтобы какой-то важный параметр вёл себя не монотонно, т.е. сначала рос, а после прохождения критического значения температуры или давления уменьшался. Возникло предположение, что на самом деле вода - это не единая жидкость, а смесь двух компонентов, которые различаются свойствами, например плотностью и вязкостью, а следовательно, и структурой. Такие идеи стали возникать в конце XIX века, когда накопилось много данных об аномалиях воды.
Первым идею о том, что вода состоит из двух компонентов, высказал Уайтинг в 1884 году. Его авторство цитирует Э.Ф. Фрицман в монографии "Природа воды. Тяжёлая вода", изданной в 1935 году. В 1891 году В. Ренгтен ввёл представление о двух состояниях воды, которые различаются плотностью. После неё появилось множество работ, в которых воду рассматривали как смесь ассоциатов разного состава ("гидролей").
Когда в 20-е годы определили структуру льда, оказалось, что молекулы воды в кристаллическом состоянии образуют трёхмерную непрерывную сетку, в которой каждая молекула имеет четырёх ближайших соседей, расположенных в вершинах правильного тетраэдра. В 1933 году Дж. Бернал и П. Фаулер предположили, что подобная сетка существует и в жидкой воде. Поскольку вода плотнее льда, они считали, что молекулы в ней расположены не так, как во льду, то есть подобно атомам кремния в минерале тридимите, а так, как атомы кремния в более плотной модификации кремнезёма - кварце. Увеличение плотности воды при нагревании от 0 до 4°C объяснялось присутствием при низкой температуре тридимитовой компоненты. Таким образом, модель Бернала - Фаулера сохранила элемент двухструктурности, но главное их достижение - идея непрерывной тетраэдрическои сетки. Тогда появился знаменитый афоризм И. Ленгмюра: „Океан - одна большая молекула". Излишняя конкретизация модели не прибавила сторонников теории единой сетки.
Только в 1951 году Дж. Попл создал модель непрерывной сетки, которая была не так конкретна, как модель Бернала - Фаулера. Попл представлял воду как случайную тетраэдрическую сетку, связи между молекулами в которой искривлены и имеют различную длину. Модель Попла объясняет уплотнение воды при плавлении искривлением связей. Когда в 60-70-е годы появились первые определения структуры льдов II и IX, стало ясно, как искривление связей может приводить к уплотнению структуры. Модель Попла не могла объяснить немонотонность зависимости свойств воды от температуры и давления так хорошо, как модели двух состояний. Поэтому идею двух состояний ещё долго разделяли многие учёные.
Но во второй половине XX века нельзя было так фантазировать о составе и строении „гидролей", как это делали в начале века. Уже было известно, как устроен лёд и кристаллогидраты, и многое знали про водородную связь. Помимо „континуальных" моделей (модель Попла), возникли две группы „смешанных" моделей: кластерные и клатратные. В первой группе вода представала в виде кластеров из молекул, связанных водородными связями, которые плавали в море молекул, в таких связях не участвующих. Модели второй группы рассматривали воду как непрерывную сетку (обычно в этом контексте называемую каркасом) водородных связей, которая содержит пустоты; в них размещаются молекулы, не образующие связей с молекулами каркаса. Нетрудно было подобрать такие свойства и концентрации двух микрофаз кластерных моделей или свойства каркаса и степень заполнения его пустот клатратных моделей, чтобы объяснить все свойства воды, в том числе и знаменитые аномалии.
Среди кластерных моделей наиболее яркой оказалась модель Г. Немети и Х. Шераги: предложенные ими картинки, изображающие кластеры связанных молекул, которые плавают в море несвязанных молекул, вошли во множество монографий.
Первую модель клатратного типа в 1946 году предложил О.Я. Самойлов: в воде сохраняется подобная гексагональному льду сетка водородных связей, полости которой частично заполнены мономерными молекулами. Л. Полинг в 1959 году создал другой вариант, предположив, что основой структуры может служить сетка связей, присущая некоторым кристаллогидратам.
В течение второй половины 60-х годов и начала 70-х наблюдается сближение всех этих взглядов. Появлялись варианты кластерных моделей, в которых в обеих микрофазах молекулы соединены водородными связями. Сторонники клатратных моделей стали допускать образование водородных связей между пустотными и каркасными молекулами. То есть фактически авторы этих моделей рассматривают воду как непрерывную сетку водородных связей. И речь идёт о том, насколько неоднородна эта сетка (например, по плотности). Представлениям о воде как о водородно-связанных кластерах, плавающих в море лишённых связей молекул воды, был положен конец в начале восьмидесятых годов, когда Г. Стэнли применил к модели воды теорию перколяции, описывающую фазовые переходы воды.
В 1999 г. известный российский исследователь воды С.В. Зенин защитил в Институте медико-биологических проблем РАН докторскую диссертацию, посвященную кластерной теории, которая явилась существенным этапом в продвижении этого направления исследований, сложность которых усиливается тем, что они находятся на стыке трех наук: физики, химии и биологии. Им на основании данных, полученных тремя физико-химическими методами: рефрактометрии (С.В. Зенин, Б.В. Тяглов, 1994), высокоэффективной жидкостной хроматографии (С.В. Зенин с соавт., 1998) и протонного магнитного резонанса (С.В. Зенин, 1993) построена и доказана геометрическая модель основного стабильного структурного образования из молекул воды (структурированная вода), а затем (С.В. Зенин, 2004) получено изображение с помощью контрастно-фазового микроскопа этих структур.
Сейчас наукой доказано, что особенности физических свойств воды и многочисленные короткоживущие водородные связи между соседними атомами водорода и кислорода в молекуле воды создают благоприятные возможности для образования особых структур-ассоциатов (кластеров), воспринимающих, хранящих и передающих самую различную информацию.
Структурной единицей такой воды является кластер, состоящий из клатратов, природа которых обусловлена дальними кулоновскими силами. В структуре кластров закодирована информация о взаимодействиях, имевших место с данными молекулами воды. В водных кластерах за счёт взаимодействия между ковалентными и водородными связями между атомами кислорода и атомами водорода может происходить миграция протона (Н+) по эстафетному механизму, приводящие к делокализации протона в пределах кластера.
Вода, состоящая из множества кластеров различных типов, образует иерархическую пространственную жидкокристаллическую структуру, которая может воспринимать и хранить огромные объемы информации.
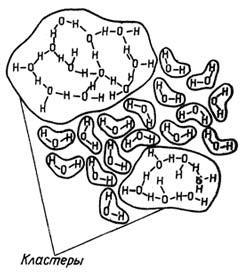
На рисунке (В.Л. Воейков) в качестве примера приведены схемы нескольких простейших кластерных структур.
Некоторые возможные структуры кластеров воды
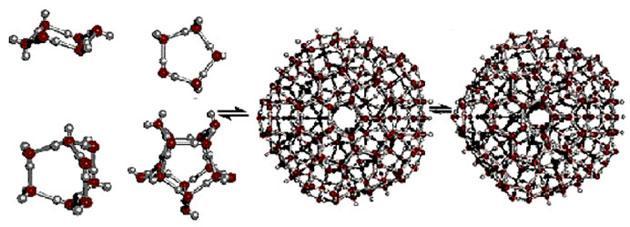
Переносчиками информации могут быть физические поля самой различной природы. Так установлена возможность дистанционного информационного взаимодействия жидкокристаллической структуры воды с объектами различной природы при помощи электромагнитных, акустических и других полей. Воздействующим объектом может быть и человек.
Вода является источником сверхслабого и слабого переменного электромагнитного излучения. Наименее хаотичное электромагнитное излучение создаёт структурированная вода. В таком случае может произойти индукция соответствующего электромагнитного поля, изменяющего структурно-информационные характеристики биологических объектов.
В течение последних лет получены важные данные о свойствах переохлаждённой воды. Изучать воду при низкой температуре очень интересно, поскольку её удаётся сильнее переохладить, чем другие жидкости. Кристаллизация воды, как правило, начинается на каких-то неоднородностях - либо на стенках сосуда, либо на плавающих частичках твердых примесей. Поэтому найти температуру, при которой бы переохлаждённая вода самопроизвольно закристаллизовалась нелегко. Но учёным удалось это сделать, и сейчас температура так называемой гомогенной нуклеации, когда образование кристаллов льдов идёт одновременно по всему объёму, известна для давлений вплоть до 0,3 ГПа, то есть захватывая области существования льда II.
От атмосферного давления до границы, разделяющей льды I и II, эта температура падает от 231 до 180 К, а потом слегка увеличивается - до 190К. Ниже этой критической температуры жидкая вода невозможна в принципе.
Однако с этой температурой связана одна загадка. В середине восьмидесятых годов была открыта новая модификация аморфного льда - лёд высокой плотности, и это помогло возрождению представлений о воде как о смеси двух состояний. В качестве прототипов рассматривались не кристаллические структуры, а структуры аморфных льдов разной плотности. В наиболее внятном виде эту концепцию сформулировали Е.Г. Понятовский и В.В. Синицин, которые в 1999 году написали: „Вода рассматривается как регулярный раствор двух компонентов, локальные конфигурации в которых соответствуют ближнему порядку модификаций аморфного льда". Более того, изучая ближний порядок в переохлаждённой воде при высоком давлении методами дифракции нейтронов, учёным удалось найти компоненты, соответствующие этим структурам.
Следствием полиморфизма аморфных льдов стали также предположения о расслоении воды на два несмешивающихся компонента при температуре ниже гипотетической низкотемпературной критической точки. К сожалению, по оценке исследователей, эта температура при давлении 0,017 ГПа равна 230К - ниже температуры нуклеации, поэтому наблюдать расслоение жидкой воды никому ещё не удалось. Так возрождение модели двух состояний поставило вопрос о неоднородности сетки водородных связей в жидкой воде. Разобраться в этой неоднородности можно только с помощью компьютерного моделирования.
Говоря о кристаллической структуре воды, следует отметить, что известно 14 модификаций льда, большинство из которых не встречаются в природе, в которых молекулы воды и сохраняют свою индивидуальность, и соединены водородными связями. С другой стороны существует множество вариантов сетки водородных связей в клатратных гидратах. Энергии этих сеток (льдов высокого давления и клатратных гидратов) ненамного выше энергий кубического и гексагонального льдов. Поэтому фрагменты таких структур также могут появляться в жидкой воде. Можно сконструировать бесчисленное множество различных непериодических фрагментов, молекулы в которых имеют по четыре ближайших соседа, расположенных приблизительно по вершинам тетраэдра, но при этом их структура не соответствует структурам известных модификаций льда. Как показали многочисленные расчёты, энергии взаимодействия молекул в таких фрагментах будут близки друг к другу, и нет оснований говорить, что какая-то структура должна преобладать в жидкой воде.
Структурные исследования воды можно изучать разными методами; спектроскопией протонного магнитного резонанса, инфракрасной спекроскопии, дифракцией рентгеновских лучей и др. Например, дифракцию рентгеновских лучей и нейтронов в воде изучали много раз. Однако подробных сведений о структуре эти эксперименты дать не могут. Неоднородности, различающиеся по плотности, можно было бы увидеть по рассеянию рентгеновских лучей и нейтронов под малыми углами, однако такие неоднородности должны быть большими, состоящими из сотен молекул воды. Можно было бы их увидеть, и исследуя рассеяние света. Однако вода - исключительно прозрачная жидкость. Единственный же результат дифракционных экспериментов - функции радиального распределения, то есть расстояния между атомами кислорода, водорода и кислорода-водорода. Из них видно, что никакого дальнего порядка в расположении молекул воды нет. Эти функции для воды затухают гораздо быстрее, чем для большинства других жидкостей. Например, распределение расстояний между атомами кислорода при температуре, близкой к комнатной, даёт только три максимума, на 2,8, 4,5 и 6,7 Å. Первый максимум соответствует расстоянию до ближайших соседей, и его значение примерно равно длине водородной связи. Второй максимум близок к средней длине ребра тетраэдра - вспомним, что молекулы воды в гексагональном льду располагаются по вершинам тетраэдра, описанного вокруг центральной молекулы. А третий максимум, выраженный весьма слабо, соответствует расстоянию до третьих и более далёких соседей по водородной сетке. Этот максимум и сам не очень ярок, а про дальнейшие пики и говорить не приходится. Были попытки получить из этих распределений более детальную информацию. Так в 1969 году И.С. Андрианов и И.З. Фишер нашли расстояния вплоть до восьмого соседа, при этом до пятого соседа оно оказалось равным 3 Å, а до шестого - 3,1 Å. Это позволяет делать данные о дальнем окружении молекул воды.
Другой метод исследования структуры - нейтронная дифракция на кристаллах воды осуществляется точно также, как и рентгеновская дифракция. Однако из-за того, что длины нейтронного рассеяния различаются у разных атомов не столь сильно, метод изоморфного замещения становится неприемлемым. На практике обычно работают с кристаллом, у которого молекулярная структура уже приблизительно установлена другими методами. Затем для этого кристалла измеряют интенсивности нейтронной дифракции. По этим результатам проводят преобразование Фурье, в ходе которого используют измеренные нейтронные интенсивности и фазы, вычисляемые с учётом неводородных атомов, т.е. атомов кислорода, положение которых в модели структуры известно. Затем на полученной таким образом фурье-карте атомы водорода и дейтерия представлены с гораздо большими весами, чем на карте электронной плотности, т.к. вклад этих атомов в нейтронное рассеяние очень большой. По этой карте плотности можно, например, определить положения атомов водорода (отрицательная плотность) и дейтерия (положительная плотность).
Возможна разновидность этого метода, которая состоит в том, что кристалл образовавшийся в воде, перед измерениями выдерживают в тяжёлой воде. В этом случае нейтронная дифракция не только позволяет установить, где расположены атомы водорода, но и выявляет те из них, способные обмениваться на дейтерий, что особенно важно при изучение изотопного (H-D)-обмена. Подобная информация помогает подтвердить правильность установления структуры.
Другие методы также позволяют изучать динамику молекул воды. Это эксперименты по квазиупругому рассеянию нейтронов, сверхбыстрой ИК-спектроскопии и изучение диффузии воды с помощью ЯМР или меченых атомов дейтерия. Метод ЯМР-спектроскопии основан на том, что ядро атома водорода имеет магнитный момент - спин, взаимодействующий с магнитными полями, постоянными и переменными. По спектру ЯМР можно судить о том, в каком окружении эти атомы и ядра находятся, получая, таким образом, информацию о структуре молекулы.
В результате экспериментов по квазиупругому рассеянию нейтронов в кристаллах воды был измерен важнейший параметр - коэффициент самодиффузии при различных давлениях и температурах. Чтобы судить о коэффициенте самодиффузии по квазиупругому рассеянию нейтронов, необходимо сделать предположение о характере движения молекул. Если они движутся в соответствии с моделью Я.И. Френкеля (известного отечественного физика-теоретика, автора „Кинетической теории жидкостей" - классической книги, переведённой на многие языки), называемой также моделью „прыжок-ожидание", тогда время „осёдлой" жизни (время между прыжками) молекулы составляет 3,2 пикосекунды. Новейшие методы фемтосекундной лазерной спектроскопии позволили оценить время жизни разорванной водородной связи: протону требуется 200 фс для того, чтобы найти себе партнёра. Однако всё это средние величины. Изучить детали строения и характера движения молекул воды можно только при помощи компьютерного моделирования, называемого иногда численным экспериментом.

Так выглядит структура воды по результатам компьютерного моделирования (по данным д.х.н. Г. Г. Маленкова). Общую беспорядочную структуру можно разбить на два типа областей (показаны тёмными и светлыми шариками), которые различаются по своему строению, например по объёму многогранника Вороного (а), степени тетраэдричности ближайшего окружения (б), значению потенциальной энергии (в), а также по наличию четырёх водородных связей у каждой молекулы (г). Впрочем, эти области буквально через мгновение, спустя несколько пикосекунд, изменят свое расположение.
Моделирование проводится так. Берётся структура льда и, нагревается до расплавления. Затем после некоторого времени, чтобы вода "забыла" о кристаллическом происхождении, снимаются мгновенные микрофотографии.
Для анализа структуры воды выбираются три параметра: - степень отклонения локального окружения молекулы от вершин правильного тетраэдра; -потенциальная энергия молекул; -объём так называемого многогранника Вороного.
Чтобы построить этот многогранник, берут ребро от данной молекулы до ближайшей, делят его пополам и через эту точку проводят плоскость, перпендикулярную ребру. Получается объём, приходящийся на одну молекулу. Объём полиэдра - это плотность, тетраэдричность - степень искажения водородных связей, энергия - степень устойчивости конфигурации молекул. Молекулы с близкими значениями каждого из этих параметров стремятся сгруппироваться вместе в отдельные кластеры. Области как с низкой, так и с высокой плотностью обладают разными значениями энергии, но могут иметь и одинаковые значения. Эксперименты показали, что области с разным строением кластеры возникают спонтанно и спонтанно распадаются. Вся структура воды живёт и постоянно меняется, причём время, за которое происходят эти изменения, очень маленькое. Исследователи следили за перемещениями молекул и выяснили, что они совершают нерегулярные колебания с частотой около 0,5 пс и амплитудой 1 ангстрем. Наблюдались также и редкие медленные скачки на ангстремы, которые длятся пикосекунды. В общем, за 30 пс молекула может сместиться на 8-10 ангстрем. Время жизни локального окружения тоже невелико. Области, составленные из молекул с близкими значениями объёма многогранника Вороного, могут распасться за 0,5 пс, а могут жить и несколько пикосекунд. А вот распределение времён жизни водородных связей очень велико. Но это время не превышает 40 пс, а среднее значение - несколько пс.
В заключение следует подчеркнуть, что теория кластерного строения воды имеет много подводных камней. Например, Зенин предполагает, что основной структурный элемент воды - кластер из 57 молекул, образованный слиянием четырёх додекаэдров. Они имеют общие грани, а их центры образуют правильный тетраэдр. То, что молекулы воды могут располагаться по вершинам пентагонального додекаэдра, известно давно; такой додекаэдр - основа газовых гидратов. Поэтому ничего удивительного в предположении о существовании таких структур в воде нет, хотя уже говорилось, что никакая конкретная структура не может быть преобладающей и существовать долго. Поэтому странно, что этот элемент предполагается главным и что в него входит ровно 57 молекул. Из шариков, например, можно собирать такие же структуры, которые состоят из примыкающих друг к другу додекаэдров и содержат 200 молекул. Зенин же утверждает, что процесс трёхмерной полимеризации воды останавливается на 57 молекулах. Более крупных ассоциатов, по его мнению, быть не должно. Однако если бы это было так, из водяного пара не могли бы осаждаться кристаллы гексагонального льда, которые содержат огромное число молекул, связанных воедино водородными связями. Совершенно неясно, почему рост кластера Зенина остановился на 57 молекулах. Чтобы уйти от противоречий, Зенин и упаковывает кластеры в более сложные образования - ромбоэдры - из почти тысячи молекул, причём исходные кластеры друг с другом водородных связей не образуют. Почему? Чем молекулы на их поверхности отличаются от тех, что внутри? По мнению Зенина, узор гидроксильных групп на поверхности ромбоэдров и обеспечивает память воды. Следовательно, молекулы воды в этих крупных комплексах жёстко фиксированы, и сами комплексы представляют собой твёрдые тела. Такая вода не будет течь, а температура её плавления, которая связана с молекулярной массой, должна быть весьма высокой.
Какие свойства воды объясняет модель Зенина? Поскольку в основе модели лежат тетраэдрические постройки, её можно в той или иной степени согласовать с данными по дифракции рентгеновских лучей и нейтронов. Однако вряд ли модель может объяснить уменьшение плотности при плавлении - упаковка додекаэдров менее плотная, чем лёд. Но труднее всего согласуется модель с динамическими свойствами - текучестью, большим значением коэффициента самодиффузии, малыми временами корреляции и диэлектрической релаксации, которые измеряются пикосекундами.
Донорно-акцепторный механизм (иначе координационный механизм) — способ образования ковалентной химической связи между двумя атомами или группой атомов, осуществляемая за счет неподеленной пары электронов атома-донора и свободной орбитали атома-акцептора.
Термины «донорно-акцепторная связь» или «координационная связь» некорректны, поскольку это не есть вид химической связи, а лишь теоретическая модель, описывающая особенность её образования. Свойства ковалентной химической связи, образованной по донорно-акцепторному механизму, ничем не отличаются от свойств связей, образованных по обменному механизму (например, связи N—H в ионе аммония NH4+ или связи O—H в ионе гидроксония Н3O+).

![]()
Образование аддукта аммиака и трифторида бора
Донорами обычно выступают атомы азота, кислорода, фосфора, серы и др., имеющие неподелённые электронные пары на валентных орбиталях малого размера. Роль акцептора могут выполнять ионизированный атом водорода H+, некоторые p-металлы (напр., алюминий при образовании иона AlH4-) и, в особенности, d-элементы, имеющие незаполненные энергетические ячейки в валентном электронном слое.
Именно с позиций донорно-акцепторного механизма описывается образование локализованных ковалентных связей в молекулах и молекулярных ионах комплексных (координационных) соединений: связь формируется за счёт неподелённой пары электронов лиганда и свободной орбитали атома-комплексообразователя. Донорно-акцепторный механизм также описывает образование промежуточных продуктов (интермедиатов) реакции, например, комплексов с переносом заряда.
Модель донорно-акцепторного механизма существуют только в рамках представлений о валентности как о локализации электронной плотности при образовании ковалентных связей (метод валентных схем). В рамках метода молекулярных орбиталей необходимости в подобных представлениях нет.
Образование комплексных соединений
Комплексные соединения имеют сложный состав. В них выделяют центральный атом (комплексообразователь) и связанные с ним лиганды (заряженные или нейтральные неорганические или органические частицы, т. е. ионы или молекулы). Комплексообразователь вместе с лигандами составляет внутреннюю координационную сферу соединения. Кроме внутренней сферы, в большинстве случаев имеется также внешняя координационная сфера, состоящая из отрицательных или положительных ионов.
Ионы внешней сферы связаны с комплексным ионом ионогенно, т. е. при растворении вещества в воде они отщепляются в виде свободных ионов. Наоборот, связь между комплексообразователем и лигандами имеет неионогенный характер, и вся внутренняя координационная сфера находится в водных растворах в виде комплексного иона. Так, в случае K[Ag(CN)2] комплексообразователем является Ag+, лигандами – CN-. Во внешней координационной сфере находятся К+ - ионы. Группа является внутренней сферой комплекса.
Заряды комплексных ионов равняются алгебраической сумме зарядов комплексообразователя и лигандов. Так, заряд иона равен (+1) + (-2) = -1.
Если лигандами являются нейтральные молекулы, их присутствие не отражается на заряде комплекса. Поэтому его заряд равен в таких случаях заряду комплексообразователя. Например, заряд иона [Ag(NH3) 2]+ такой же, как и у иона Ag+.
О заряде комплекса можно судить также по составу внешней координационной сферы. Найдя заряд комплекса, легко определить и степень окисления комплексообразователя, если она неизвестна. Так, в комплексном соединении K4 [Fe(CN)6] во внешней сфере находятся четыре однозарядных положительных иона К+, а поскольку вся молекула электронейтральна, заряд внутренней координационной сферы [Fe(CN)6]4- должен равняться 4-. Ионы CN- , являющиеся лигандами, несут в общей сложности 6 отрицательных зарядов, значит, комплексо-образователь должен иметь заряд, равный 2+.
Общее число лигандов, координированных с данным комплексообразователем в комплекс, называется координационным числом комплексообразователя. У большинства комплексообразователей (Cr3+, Co3+, Fe2+, Fe 3+, Zn2+, Ni2+ и т. д.) координационное число равно 6. Координационное число 4 наблюдается у ионов Cd2+, Cu2+, Hg2+, Pt2+ и др. Существуют также комплексы, в которых комплексообразователь может иметь и другие координационные числа, например: 2, 3, 8 и т. д.
Лигандами в большинстве случаев служат однозарядные отрицательные ионы, а также молекулы, подобные NH3, H2O, и другие, способные занимать лишь одно координационное место в комплексе. Известны лиганды, которые могут занимать сразу два и больше мест. Такие лиганды называются бидентатными, тридентатными и т. д., в зависимости от того, сколько мест во внутренней сфере комплексного соединения данный лиганд может занять.
Способность к образованию комплексов всего сильнее проявляют элементы, расположенные в средней части больших периодов Периодической системы элементов Д. И. Менделеева (Cu, Ni, Co, Fe, Mn, Zn, Ag и т. д.).
Общим методом получения комплексов типа присоединения является действие избытка комплексообразующего реагента на соли связываемого в комплекс иона. Например,
HgCl2 + 4KI = K2[HgI4] + 2KCl
Таким способом получают галогено-, родано-, циано-, нитро-, киcлородсодержащие и другие комплексные соединения.
Общим методом получения комплексов, образующихся по типу внедрения, является действие аммиака или воды на соли связываемого в комплекс иона. Например,
HgCl2 + 2NH4OH = [Ag(NH3)2]Cl + 2H2O
Так получают аммиакаты и аквакомплексы.
С позиций электронной теории образование комплексных соединений происходит как результат проявления сил электростатического притяжения, действующих между лигандами и ионом-комплексообразователем.
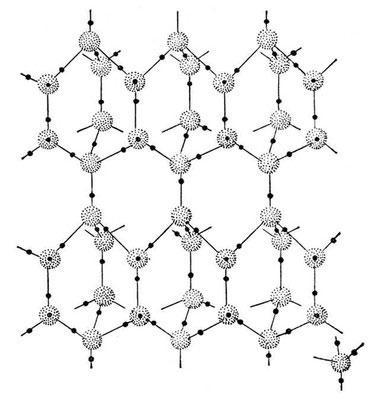
Электростатическая теория Косселя (теория ионной связи) образование молекулы аммиака объясняет следующим образом. Нейтральный атом азота во внешнем электронном слое имеет 5 электронов и поэтому стремится дополнить это число до устойчивого октета (8 электронов). При реакции с атомами водорода электроны трех атомов водорода переходят к атому азота и образуется отрицательный N3- и три положительных H+. В результате взаимного притяжения образуется [NH3].
Теория ковалентной связи Льюиса образование комплексов объясняет возникновением ковалентной связи между ионами комплексообразователя и лигандами. Ковалентная связь осуществляется за счет электронных пар, общих для обоих соединяющихся атомов. Согласно этой теории молекула аммиака образуется в результате возникновения трех общих электронных пар, связывающих атомы водорода с атомом азота:
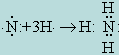
Теория ковалентной связи допускает возможность использования "свободной" пары электронов для осуществления связи с соответствующим ионом. В случае аммиака таким ионом является H+, в результате образуется ион NH+4:
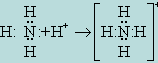
Подобная связь, осуществляемая за счет электронной пары, принадлежавшей первоначально одному из соединяющихся атомов и как бы отдаваемой им в общее владение с другим атомом, называется координационной, или донорно-акцепторной связью. При этом атом, отдающий электронную пару, называют донором, а атом, получающий её,– акцептором.
Лиганды в комплексных соединениях в большинстве случаев представляют собой атомы или группы атомов с одной или несколькими неподеленными парами электронов. К ним относятся галогенид-ионы, азотистые основания (аммиак, пиридин, алифатические амины и др.), гидроксил-ионы, некоторые органические соединения, содержащие карбоксильную (СООН- ) и карбонильную (= С = О) группы, а также функциональные группы, имеющие в своем составе серу, фосфор или мышьяк. Донорными атомами в этих лигандах, как правило, являются галоген, азот, кислород, фосфор или мышьяк.
Следующим этапом развития теории координационных соединений являются теория молекулярных орбиталей, которая развивает и углубляет теорию ковалентных связей, и теория строения кристаллического поля, которая основывается на электрохимических представлениях. Более полной теорией координационных соединений, объединяющей теорию молекулярных орбиталей и теорию кристаллического поля, является теория поля лигандов.
Устойчивость комплексных соединений. Константа нестойкости
В растворах комплексных соединений существует система динамических равновесий, т. е. комплексные соединения подвержены в значительной степени электролитической диссоциации. Ионы или молекулы, находящиеся во внешней сфере, связаны в комплексном соединении гораздо слабее, чем ионы или молекулы, находящиеся во внутренней сфере. Такая различная прочность связи обусловливает характер диссоциации комплексных соединений.
Диссоциация комплексных соединений протекает по стадиям. На первой стадии комплексное соединение диссоциирует как сильный электролит, т. е. практически полностью. Далее, комплексный ион диссоциирует как слабый электролит по ступеням, т. е. лиганды отделяются от комплексообразователя постепенно. Например, диссоциация комплексной соли [Ag(NH3)2]Cl протекает по следующим стадиям:
Первая
стадия:
[Ag(NH3)2]Cl
![]() [Ag(NH3)2]+
+ Cl-
[Ag(NH3)2]+
+ Cl-
Вторая
стадия:1.
Ag(NH3)2]+
![]() AgNH3+
+ NH3
AgNH3+
+ NH3
![]()
2.
AgNH3+![]() Ag+
+ NH3
Ag+
+ NH3
![]()
Диссоциация комплекса идет лишь в незначительной степени и может быть охарактеризована величиной общей константы ионизации данного комплекса, которая определяется как произведение констант диссоциации по отдельным ступеням. Так, в данном примере
![]() (9.1)
(9.1)
Чем больше величина этой константы, тем сильнее комплекс диссоциирует, тем менее он устойчив. Эта константа называется константой нестойкости. Обратная величина константы нестойкости называется константой устойчивости :
![]() (9.2)
(9.2)
Константы нестойкости для различных комплексных ионов различны и могут служить мерой устойчивости комплекса. Наиболее устойчивые в растворах комплексные ионы имеют наименьшие константы нестойкости. Так, среди соединений
|
|
[Ag(NO2)2] - |
[Ag(NH3)2] + |
[Ag(S2O3)] - |
[Ag(CN)2] - |
|
K нест |
1,3 · 10-3 |
5,8 · 10 -8 |
1,5 · 10 -9 |
1,4 · 10-20 |
устойчивость комплекса возрастает при переходе от [Ag(NO2)2] - к [Ag(CN)2] -.
Значения констант нестойкости и устойчивости приводятся в справочниках по химии. С помощью этих величин можно предсказать течение реакций между комплексными соединениями: при сильном различии констант устойчивости реакция пойдет в сторону образования комплекса с большей константой устойчивости или, что равноценно, с меньшей константой нестойкости. Например, для иона [Ag(NH3)2] + Kнест = 5,8 · 10 -8, а для иона [Ag(CN)2] - Kнест = 1,4 · 10 -20, поэтому при действии раствора КСN аммиакат серебра разрушается с образованием иона [Ag(CN)2] - :
[Ag(NH3)2]Cl = 2KCN = K[Ag(CN)2] + 2NH3 = KCl
Зная константы нестойкости различных комплексов, можно теоретически вычислить концентрацию соответствующих простых ионов в растворах комплексных солей.
Пример 1. Найти концентрацию Ag+ в 0,01 М растворе [Ag(NH3)2]NO3, если Kнест = 5,8 · 10 - 8.
Решение. Обозначив искомую концентрацию Ag+ через х, из уравнения диссоциации комплекса [Ag(NH3)2]+ находим
[NH3]
= 2x;
![]()
т. к. степень диссоциации комплекса незначительна. Следовательно, можно записать
![]()
![]()
От величины Kнест комплексного иона и KS осадка, который образует реактив с одним из продуктов распада комплекса, зависит, будут ли образовываться соответствующие осадки.
Пример 2. Образуется ли осадок AgCl при прибавлении к 0,01М раствору [Ag(NH3)2]NO3 0,01М раствора KСl?
KS(AgCl) = 1,8 · 10 -10; Kнест[Ag(NH3)+2] = 5,8 10 -8.
Решение. [ Ag+] = 5,25 ·10 -4 моль/л (см. пример 1), исходя из условия [Cl]- = 0,01 моль/л.
Для того чтобы выпал осадок AgCl, необходимо, чтобы
[ Ag+] · [Cl-]>KS(AgCl)
В данном примере
5,25 · 10 -4 · 10-2 = 5,25 · 10 -6 > 1,8 · 10 -10
Следовательно, осадок AgCl образуется.
При выборе условий наиболее эффективного разделения ионов исходят из соотношения констант устойчивости образуемых ими комплексных соединений.
Например, катионы Ni2+, Co2+, Zn2+ дают устойчивые растворимые аммиакаты, а Al3+, Fe3+, Cr3+ менее склонны к комплексообразованию с аммиаком и осаждаются при действии аммиака в виде гидроксидов. Это позволяет разделить действием аммиака эти две группы катионов.
Внутрикомплексные соединения
Комплексные соединения, образуемые различными катионами с органическими реагентами, относятся к типу внутрикомплексных солей и имеют большое значение в аналитической химии. Причина этого – их большая устойчивость по сравнению с неорганическими комплексами и более интенсивная окраска.
Основоположниками применения органических аналитических реактивов являются М. А. Ильинский, применивший α-нитрозо- β-нафтол для обнаружения кобальта, и Л. А. Чугаев, синтезировавший и применивший в анализе диметилглиоксим для обнаружения и количественного определения никеля. Координационная связь обозначена стрелкой, направленной к центральному атому-комплексообразователю. Подобной внутриком-плексной солью является соединение Ni2+ с диметилглиоксимом (рис. 5.3.1.). Для образования внутрикомплексных соединений требуется участие органических реагентов, которые содержат определенные атомные группы, обладающие кислотными свойствами. Такими группами являются: -COOH, -OH, =NOH, =NH, -NH2, -SO3H и др. Ион-комплексообразователь может вытеснять водород из таких групп органического соединения, образуя обычную валентную связь. Если же в молекуле органического соединения, наряду с одной из подобных групп, присутствует другая группа, играющая роль лиганда для данного катиона (-NH2, =NH, =CO, -OH, =S и др.), то катион, замещая атом водорода в кислотной группе, может образовывать координационную связь с группой лиганда. Получающиеся при этом соли называются внутрикомплексными. Внутрикомплексные соединения – соединения, в которых лиганд связан с комплексообразователем одновременно ионными и координационными связями. Например, при взаимодействии аминоуксусной кислоты NH2 - CH2 - COOH с ионами Cu2+ образуется соединение циклической структуры:
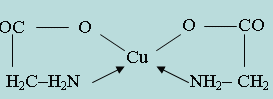
Координационная связь обозначена стрелкой, направленной к центральному атому-комплексообразователю. Подобной внутрикомплексной солью является соединение Ni2+ с диметилглиоксимом (рис. 9.3).
Таким образом, наряду с замещением двух атомов водорода в кислотных группах = NOH двух молекул диметилглиоксима, Ni2+-ион соединяется также координационной связью с атомами азота еще двух указанных групп. Из приведенных примеров видно, что молекулы внутрикомплексных солей имеют циклическое (кольцеобразное) строение. Комплексные соединения, имеющие циклическую структуру, называют хелатами. Частным случаем их являются внутрикомплексные соединения. Наиболее устойчивые внутрикомплексные соединения с пяти- и шестичленными кольцами.
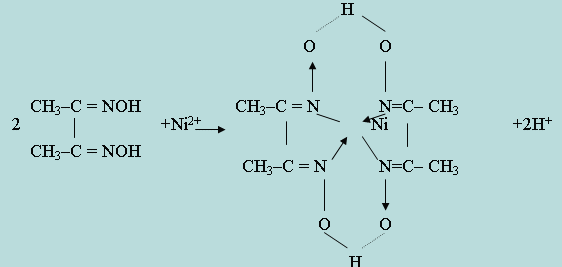
Рис.9.3 Уравнение реакции образования никеля с диметилглиоксимом
Широкое применение в практике химического анализа получили органические вещества, образующие с большинством катионов прочные растворимые комплексы. Эти вещества называются комплексонами. Комплексоны представляют собой многоосновные органические кислоты (или их соли), содержащие аминогруппы. Например, этилендиаминтетрауксусная кислота:
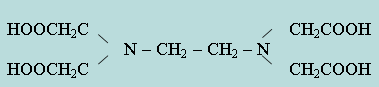
Двунатриевая соль этой кислоты, или комплексон III (трилон Б), образует растворимые комплексы со всеми двух-, трех- и четырехзарядными катионами при различных рН раствора. Большое применение комплексоны получили в количественном анализе.
Значение комплексных соединений в химическом анализе
Реакции комплексообразования играют важную роль во многих аналитических процессах. Комплексообразующие реагенты часто добавляют, чтобы предотвратить нежелательные реакции. Например, при осаждении никеля диметилглиоксимом в раствор добавляют соль винной кислоты, образующей комплекс с железом (3+) и удерживающей его в растворе. Многие комплексы с органическими реагентами нерастворимы в воде и их используют для разделения или гравиметрического определения ионов металла. При количественном спектрофотометриче-ском определении различных элементов большое значение имеют реагенты, образующие с этими элементами ярко окрашенные комплексы. Во многих случаях за счет избирательного комплексообразования осуществляется ионообменное разделение, а в основе экстракционного разделения часто лежит экстракция комплексов.
Реакции образования комплексов в химическом анализе используются для количественного определения ионов методом комплексо-метрического титрования.