

Геохімія
.pdf121
6.7 Застосування стабільних ізотопів при вивченні генезису магми
Розподіл стабільних ізотопів в процесі магматичної диференціації відбувається під дією кількох чинників, до яких належать зокрема: тип і послідовність кристалізації; ефективність фракціонування. Співвідношення ізотопів кисню при диференціації основної магми (за виключенням останніх стадій) змінюється лише незначним чином, що обумовлено високою температурою кристалізації. Тенденція до зростання величини 18O відчутно проявляється при зростанні вмісту кремнезему, тому незмінені ультраосновні породи мають 18O=+5 +7‰, габро, базальти, андезити і сієніти - 18O=+5,5 +7,5‰, а граніти - 18O=+7 +13‰.
Згідно з даними Тейлора, який опублікував результати ізотопного аналізу великої кількості магматичних порід і мінералів з різних регіонів планети, більшість вулканічних та інтрузивних порід виявились дуже однорідними як за 18O (+5,5 +10‰), так і за D (-50 -80‰). Така однорідність обумовлена спільністю джерел магми та існуючими тісними взаємозв’язками (виникають при кристалізаційній диференціації) магматичних порід різних типів. Фактично стабільні ізотопи надають небагато інформації щодо особливостей петрогенезису породи для якої характерні звичайні співвідношення ізотопів, і можуть використовуватись для виявлення випадків відхилення розподілу ізотопів від норми для порід цього типу.
Для порід з аномальними величинами ізотопних співвідношень вірогідними є або незвичайні джерела, або гідротермальні зміни.
Враховуючи тенденцію збагачення сіалічної кори 18O (порівняно з породами мантійного походження), можна припускати що магматичні породи з аномально високим вмістом 18O утворювались або в результаті плавлення, або в результаті асиміляції збагаченого 18O сіалічного матеріалу. Так, в магматичній провінції Тоскана (Італія) високі значення 18O (+15,3 +16,4‰) в ріолітах і дацитах можуть бути пояснені лише за припущення що їх утворення пов’язане з поавленням чи значною асиміляцією збагачених 18O глинистих сланців (це підтверджується даними по ізотопії стронцію - 87Sr/86Sr=0,713-0,720 та присутністю кордієриту).
Вплив послідовності кристалізації на співвідношення ізотопів можна проілюструвати наступним чином: рання кристалізація магнетиту призводить до збагачення залишкової магми 18O (порівняно з такою магмою, де у зв’язку з низькою летючістю кисню магнетит не утворюється).
Існують формули для оцінки тенденцій фракціонування ізотопів при кристалізації магми. Зокрема для моделі Релея:
Rl/RO=[f+ (1-f)]-1, де =RS/Rl |
(6.16) |
при цьому Rl – співвідношення ізотопів в розплаві, RS – співвідношення ізотопів в кристалізованій речовині, RO – співвідношення ізотопів в вихідній магмі, f - частка залишкового розплаву.
Це рівняння легко реалізувати використавши дані про валовий склад породи, припустивши, при цьому, що концентрація кисню в магмі відповідає його концентрації в породі (як вважається таке припущення вносить похибку не більш як 10%). Рівняння (6.16) більш зручно виразити в величинах :
|
= ЗАЛ - O {[f+ (1-f)]-1 –1}*1000 |
(6.17) |
де ЗАЛ |
- величина параметру в магмі після кристалізації частки речовини f-1, а O = відповідає |
|
первинній магмі. |
|
|
Для силікатів величина значно менша за 0,998 є малоймовірною (тобто = 18OКРИСТ- 18OЗАЛ -2). А для =0,999, навіть після того як 99% речовини буде кристалізовано, співвідношення ізотопу в залишковому розплаві зміниться тільки на 1 тисячну.
Часткова кристалізація подібна до Релеєвської моделі часткової дистиляції, тому можна використати рівняння:
121

=1000*(f -1 –1) |
122 |
(6.18) |
Загальною рисою цих процесів є те, що продукція реакції (пара у випадку дистиляції, кристал у випадку кристалізації) тільки незначний проміжок часу перебуває в рівновазі з вихідною речовиною. В момент виникнення цей продукт вилучається з подальшої участі в реакції (тобто позбавляється можливості досягнути рівноваги з первинною фазою). Таке фракціонування найбільш помітне в вулканічних породах, однак і в цьому випадку результуючий ефект не є надто значним оскільки на загал дуже близьке до 1. На Рис.6.2 показані теоретичні криві зміни ізотопного складу розплаву при його частковій кристалізації для різних значень ( 1000( –1)). Насправді ж, змінюється під час кристалізації під впливом: (1) зміни температури; (2) зміни мінералів що кристалізуються; (3) зміни складу розплаву. Всі ці зміни загалом передбачають зростання ефективної величини в результаті кристалізації. Відповідно, ми повинні очікувати найбільшого ізотопного фракціонування в розплавах з яких кристалізуються не силікатні мінерали (наприклад магнетит) і в розплавах кристалізація яких відбувається за низьких температур (ріоліти, наприклад), а найменше фракціонування буде в розплавах які кристалізуються при високих температурах (наприклад, базальти).
Узагальнюючи дані про залежність фракціонування ізотопів кисню від температури ми можемо зробити висновок що при низьких температурах (тобто, в діапазоні температур від характерних для поверхні Землі до температур гідротермальних систем - 300-400°C), співвідношення кисневих ізотопів контролюються хімічними процесами. Величина зміни ізотопних співвідношень може бути використана як індикатор природи процесу, який був задіяний, і, за умови рівноваги,
температури за якої процес відбувався. При високих температурах (температури внутрішньої частини Землі або температури магми), співвідношення кисневих ізотопів мінімально залежать від хімічних процесів і можуть використовуватися, як індикатори (“мічені атоми”), подібно до радіогенних ізотопів.
Звідси можна сформулювати твердження:
магматичні породи, ізотопний склад кисню в яких суттєво відрізняється від вихідного значення (+6) напевно були або змінені низькотемпературними процесами, або повинні містити речовину що утворена на поверхні Землі (Taylor і Sheppard, 1986).
Рис.6.2. Залежність величини 18O від міри кристалізації розплаву згідно моделі часткової кристалізації Релея, 18O вихідної магми прийнято
+6. (за Taylor і Sheppard, 1986).
Рис.6.3. Залежність величини 18O від міри кристалізації розплаву та кількості асимільованої речовини, 18O вихідної магми прийнято +5,7. (за Taylor, 1980).
123
я визначення температурних параметрів, pH, фугітивності кисню, джерела рудоутворюючих
6.8 Застосування стабільних ізотопів при вивченні генезису рудних родовищ
Для визначення температурних параметрів, pH, фугітивності кисню, джерела рудоутворюючих флюїдів з успіхом використовують ізотопи кисню - 18O, сірки - 34S (34S/32S), і, за наявності в родовищі карбонатів або графіту, вуглецю - 13C.
За емпіричними даними загальний діапазон зміни 34SCD складає -94 +76‰, а величина цього параметру в сполуках сірки змінюється (для вірогідності 95%) в межах -40 +55‰.
Величини 34SCD в сульфідних мінералах гідротермальних родовищ змінюються в діапазоні -7 +15‰ (при загальному діапазоні для сульфідних мінералів з вірогідністю 95% - -40 +35‰)
123
124
Основною фізичною причиною фракціонування ізотопів є те що характер руху молекул (а значить і енергія речовини) залежить від мас атомів які ці молекули складають.
Відмінність мас ізотопів сприяє тому що їх розділення відбувається при обмінних реакціях,
наприклад:
HC12N + C13N- ↔ HC13N + C12N-
газ |
розчин |
газ |
розчин |
Константа хімічної рівноваги для цієї реакції дорівнює:
K = ([HC13N]*[C12N-])/([HC12N]*[C13N-]) = ([HC13N]/[HC12N]):([C13N-]/[C12N-]) =
де - коефіцієнт розділення ізотопів, який являє собою співвідношення вмістів любих двох ізотопів в хімічному з’єднанні А по відношенню до вмістів двох відповідних ізотопів в хімічному з’єднанні В: A-B = RA/RB.
Тоді коефіцієнт розділення ізотопів O18 та O16 в реакції:
H2O18 + (1/3)CaCO163 = H2O16 + (1/3)CaCO183
виражається як CaCO3H2O = (O18/O16)CaCO3 / (O18/O16)H2O = 1.031 при 25 C.
Значення звичайно близькі до 1 (1,00Х) тому прийнято використовувати для вираження коефіцієнту розділення -величини (тобто значення Х в промілях – ‰). Значення і пов’язані між собою співвідношенням: = ( -1)*1000 ‰.
Ізотопний склад двох з’єднань A і B проаналізований в лабораторії, виражається значеннями, ‰: A = (RA/RSt -1)*1000; B = (RB/RSt -1)*1000;
при цьому RSt – відоме ізотопне відношення в стандартному зразку. Тоді
A-B = ( A+1000)/ ( B+1000); або A- B =103ln( A-B).
Таблиця 2.9 – Ізотопні співвідношення C12:C13 в біосфері (за А.П.Виноградовим) – стандарт
ізотопне співвідношення морських вапняків.
Джерело вуглецю |
C12:C13 |
C13, ‰ |
CO2 атмосфери |
89,2 |
-6,6 |
CO2 океанічної води |
89,55 |
-2,0 |
Тканини морських організмів |
90,5 |
-25,0 |
Нафти і бітуми |
90,9 |
-25,0 |
Викопне вугілля |
90,78 |
-25,0 |
Морські карбонати CaCO3 |
88,5 |
0,0 |
Карбонати скелетів морських організмів |
88,7 |
0,2 |
Кам’яні метеорити |
90,9-92,5 |
-25,0 |
Згідно існуючих уявлень за ізотопним складом можна чітко розрізнити вуглець біогенного, небіогенного та метеоритного походження.
HC12N |
+ C13N- |
↔ |
HC13N |
|
|
+ C12N- |
||||
газ |
розчин |
|
газ |
|
|
розчин |
||||
Константа хімічної рівноваги для цієї реакції дорівнює: |
|
|
|
|||||||
|
K |
[HC13N]*[C12N-] |
HC13N |
|
C13N- |
|||||
|
|
|
|
|
|
: |
|
|
|
|
|
12 |
13 - |
|
12 |
12 |
- |
||||
|
|
[HC N]*[C N |
] |
HC N |
|
C N |
|
|
||
A-B = RA/RB.
124
125
Тоді коефіцієнт розділення ізотопів O18 та O16 в реакції:
H2O18 + (1/3)CaCO163 = H2O16 + (1/3)CaCO183
CaCO3-H2O =(O18/O16)CaCO3/(O18/O16)H2O= 1.031
= ( -1)*1000 ‰.
A = (RA/RSt -1)*1000; B = (RB/RSt -1)*1000;
A-B=( A+1000)/( B+1000); або A- B=103ln( A-B).
125
126
Чинники контролю розподілу елементів
Як ми вже визначали, ступінь захоплення кристалізуючимся мінералом будь-якого елементу можна виразити за допомогою коефіцієнта розподілу (k). Цей коефіцієнт відображає відношення масової концентрації елементу (M) в мінералі до його вмісту в магмі.
kM=[M]в мінералі / [M]в магмі |
8а.1 |
Значення kM може бути дуже чутливим до змін температури, тиску і складу мінералів та магми під час кристалізації (для розсіяних елементів спостерігається залежність від температури, тиску і концентрації головних елементів).
Наприклад, залежність від температури описується рівнянням типу:
lnk=A*1000/T +B |
8а.1 |
де A і B є константами ( A>+1, B – найчастіше достатньо велике від’ємне число і лише зрідка наближається до 0), а T – температура в кельвінах (наприклад коефіцієнт розподілу для стронцію в парі плагіоклаз - базальтовий розплав визначений як lnkSrPl=15121/T-9,909). Тобто з ростом температури коефіцієнт розподілу, як правило, зменшується (хоча інколи – самарій в гранаті, зростає).
Залежності коефіцієнт розподілу від складу вивчені ще недостатньо, однак відомо що розподіл нікелю в системі олівін-розплав залежить від вмісту магнію, розподіл марганцю – від відношення
Si/O.
Існує тенденція до зростання коефіцієнтів розподілу при переході від основних порід до кислих.
Для цілей петрогенетичного моделювання часто використовують валовий коефіцієнт розподілу. Так, за одночасної кристалізації мінералів Q, R і S (в пропорціях , і , при + + =1):
kM= *kMQ+ *kMR+ *kMS 8а.3
Будь який з коефіцієнтів є справедливим виключно за даних тиску і температури та рівноважного розподілу елементу між фазами.
126
127
5.2 СТРУКТУРНИЙ КОНТРОЛЬ РОЗПОДІЛУ ЕЛЕМЕНТІВ
Процеси хімічного фракціонування в кристалізуючійся магмі, незалежно від рівноважності чи нерівноважності умов, в поєднанні з протилежними процесами часткового плавлення материнських гірських порід призводять до розмаїття складів магм і широкому діапазону фракціонування великої кількості елементів. Метаморфізм та гіпергенні процеси не можуть обумовити такого ж масштабного фракціонування, однак вони можуть суттєво перерозподілити елементи в межах невеликих об’ємів гірських порід: в результаті росту нових мінералів, розчинення або перекристалізації. Однією з умов правильної постановки задачі при геохімічних дослідженнях є розуміння контролюючого впливу на розподіл елементів в породах структури
– структури розчинів, розплавів і твердих фаз. Молекулярна структура відображає спосіб заповнення і взаємне розташування атомів в хімічній сполуці і найважливіший вплив на неї мають розміри атомів, що входять до складу сполуки. Чинником, який впливає на впорядкованість кристалічної структури, є також природа хімічного зв’язку. Зокрема, в іонних твердих тілах важливе значення мають заряди іонів, оскільки їх взаємна локальна нейтралізація в кристалі та ефекти відштовхування допомагають визначити розташування в решітці іонів однакового знаку. У твердих тілах з ковалентним зв’язком важливим чинником є геометрична конфігурація локалізованого в просторі розподілу узагальнених атомами електронів.
При кристалізації відбувається обмін елементами між рідкою і твердою фазами або між твердими фазами, і відповідно, зміна хімічної стабільності при переході елементу з однієї фази в іншу. Ступінь таких енергетичних змін частково визначається структурою кристалічної речовини, розплаву, розчину.
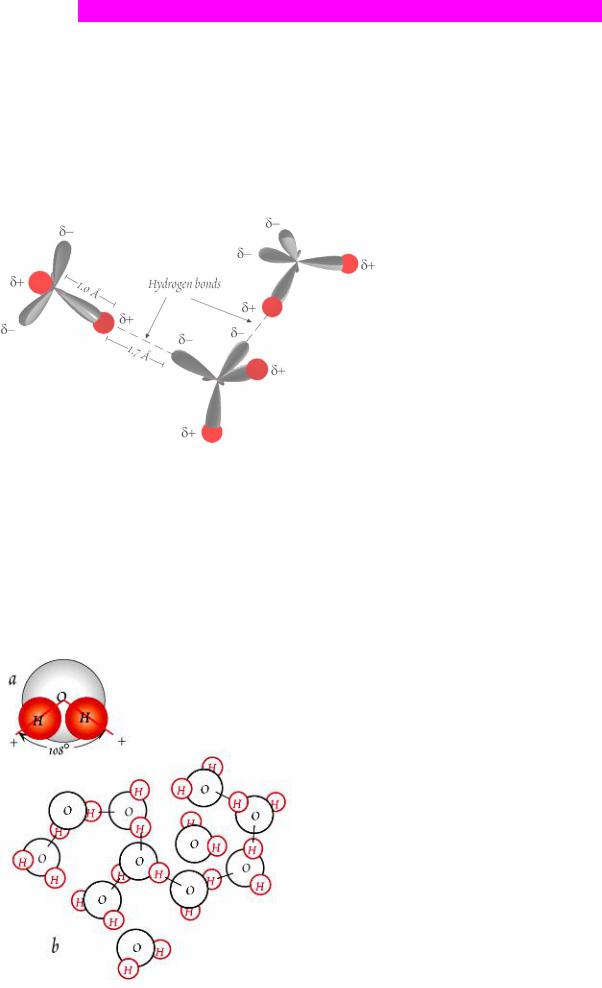
Розчини. Міжмолекулярна структура розчину значним чином обумовлена водневими зв’язками між молекулами води.
Рисунок 8.1. Водневі зв’язки між молекулами води. Червоним показані позиції водню; гібридні орбіталі sp3 в кисні показані сірим. Позначками + і + показане розташування локальних позитивних і негативних зарядів, відповідно.
Водневі зв’язки виникають, подібно до вандерваальсівських, завдяки нерівномірному (несиметричному)
розподілу заряду в молекулах, однак відрізняються шляхом реалізації. По-перше, такі зв’язки з’являються виключно між воднем і сильно електровід’ємними атомами, якими є в першу чергу кисень, а також азот і фтор. По друге, величини енергій цих взаємодій можуть бути в кілька разів вищими за вандерваальсівські, хоча й все ще меншими за енергії ковалентних та іонних зв’язків. Природа водневих зв’язків до кінця не зрозуміла, хоча ясно що виникають вони головним чином за рахунок електростатичних взаємодій. У молекулі води зв’язок між киснем і воднем передбачає гібридизацією s- і p-орбіталей, в результаті чого утворюються дві сполучні орбіталі між O і двома атомами H і 2 незв’язані sp3-орбіталі в атому кисню. Останні розташовані на протилежній від водню стороні атому O. Водень однієї молекули води, маючи позитивний заряд, притягується не зв’язаними sp3-електронами кисню іншої молекули води і формує водневий зв’язок (Рис. 8.1).
Енергія водневих зв’язків звичайно складає 20-40 kJ/mol, вона значно вища, ніж очікується у випадку електростатичних взаємодій і дійсно наближається до значень характерних для внутрішньомолекулярних зв’язків. Тому є припущення що у водневих зв’язках присутня певна ступінь ковалентності - тобто не зв’язані електрони кисню є певною мірою спільними з воднем іншої молекули. Водневі зв’язки можливо найважливіші для воді, де вони обумовлюють деякі незвичайні якості цього з’єднання, такі як висока теплота пароутворення, але вони можуть також бути важливими в органічних молекулах, в HF і аміаку.
Єдиної точки зору про структуру рідкої води на сьогодні не існує. Найбільш привабливою вважається модель викривлених водневих зв’язків, у відповідності
127
128
до якої в рідкій воді зберігається тетраедричний порядок розташування молекул і спостерігаються лише невеликі зміни відстаней між молекулами та радіусів. Розчинені у воді речовини змінюють її структуру і властивості, заповнюючи простір в середині кристалічної гратки води. Результатом є розмаїття рухливих форм розчинених у воді хімічних сполук, прояви конгруентного та інконгруентного розчинення тощо.
Рисунок 8.2. a. Структура молекули води. Кут між іонами водень-кисень водень складає 108 для води в рідкому стані і 108 у випадку водяної пари. Водень несе локальний позитивний а кисень – негативний заряд. b. Структура води в рідкому стані. Лініями показані водневі зв’язки між молекулами.
Приклади реакцій конгруентного (8.1) та інконгруентного (8.2) розчинення:
SiO2+H2O H4SiO4 8.1
кварц+вода=ортокременева кислота
MgCO3+2H2O Mg(OH)2+HCO3-+H+ 8.2
магнезит+вода=брусит+гідркарбонат+водневий іон
Кристалічна речовина. Найпоширенішими у складі земної кори та мантії є силікати. При цьому головні характеристики їх (часто досить складної) структури визначаються переважно типом з’єднання кременекисневих тетраедрів. Відмінності в ступенях зв’язку формують різні структурні типи:
-з ізольованими кременекисневими тетраедрами (олівін, в якому іони магнію та заліза розташовані таким чином, що відбувається локальна нейтралізація негативного заряду окремої групи SiO4);
-з ланцюжками тетраедрів (піроксени та амфіболи);
-з шарами тетраедрів (слюди);
-з каркасами тетраедрів, де групи (Si, Al)O4 з’єднані між собою шляхом усуспільнення атомів кисню у всіх напрямках (плагіоклази і калієві польові шпати).
Убільшості природних силікатів зв’язок між киснем тетраедрів та елементами поза тетраедрами переважно іонний, однак велика кількість можливих координаційних багатогранників з різними катіонами обумовлює широке розмаїття структур. Саме існування цих відмінних координаційних структур є одним з ключових чинників, що визначають фракціонування елементів в геохімічних процесах. Крім того, в окислах, сульфідах, самородних металах, карбонатах і силікатах спостерігаються відмінності характеру хімічного зв’язку елементів.
Вортопіроксені (Mg,Fe)2SiO6 ланцюжки складаються з кременекисневих тетраедрів, з’єднаних шляхом узагальнення двох киснів у всіх суміжних тетраедрах. Між собою ланцюжки з’єднані стрічками катіонів, які займають позиції з КЧ=6, однак ці позиції двох типів. В першій (М1) - катіон оточений шістьма атомами кисню, кожен з яких пов’язаний з одним атомом кремнію (октаедрична симетрія). В другій (М2) з шести атомів кисню чотири пов’язані з одним атомом кремнію, а два інших - є містковими, спільними для двох атомів кремнію, що порушує симетрію.
Взагальному випадку середня довжина зв’язку в М2 зростає на 2-15 %, із відхиленням кута між
зв’язками до 22 . Для Mg0,93Fe1,07SiO6 середня відстань від кисню до катіону в позиції М1 складає 210 пкм (204-217 пкм), а кут О-М1-О відхиляється від 90 не більш як на 7,3 . В позиції М2 середня довжина зв’язку складає 223 пкм (204-252 пкм), а відхилення кута О-М2-О від 90 сягає вже до 22 .
Викривлення і більший розмір позиції М2 порівняно з позицією М1 призводить до відмінності енергій зв’язку в цих позиціях, а значить і контролює перерозподіл катіонів між ортопіроксенами та іншими фазами. Явище невипадкового розподілу катіонів по наявним позиціям (катіон займає ту позицію, при якій зростає хімічна стабільність) називається явищем катіонного впорядкування. Ступінь катіонного впорядкування залежить від температури.
128

129
Рисунок 8.3. Кристалічна структура ортопіроксену. (а) Проекція (001). (б) Поліедри в проекції (100). [За Burns R.G., 1970].
(додати про шпінель)
Силікатні розплави.
Силікатні рідини відігравали, в історії розвитку Землі та інших тіл Сонячної системи, надзвичайно важливу роль. Наскільки нам відомо первісна земна кора сформувалася при підйомі розплавів до поверхні та їх охолодженні. Тому розуміння магматичних процесів є дуже важливою складовою наук про Землю. До останніх одного-двох десятиріч, основними підходами до магматичної петрології був спостережний та експериментальний. Результати експериментів з плавлення в лабораторії використовувались для інтерпретації польових спостережень. Цей підхід був дуже продуктивним і ми завдячуємо йому більшістю наших знань щодо магматичних процесів. Однак, цей підхід, як і будь-який інший, має природні межі застосування: фактично кожна магма унікальна за складом та історією кристалізації. Обмежена експериментальна база не дозволяє здійснити експерименти по плавленню кожної породи. Тому логічним, для петрологів та геохіміків що займаються вивченням магматичних процесів, є застосування термодинамічних моделей
силікатного розплаву, як інструменту інтерпретації розвитку магм. За відповідною “моделлю” взаємодії різних компонентів в силікатному розплаві і відповідними термодинамічними даними ми можемо прогнозувати стан рівноваги будь-якої магми (магма складається не лише з рідини, але й з газової та кристалічної фази) для будь-якого конкретного набору умов. Звісно, на шляху створення конкретних термодинамічних моделей розвитку магми були (і є) значні перешкоди: оскільки розплави стійкі лише за високих температур важко отримувати експериментальні дані; силікатні рідини є дуже складними комплексними розчинами, в яких в достатньо великих концентраціях (для впливу на хід реакції) знаходиться 8 або й більше компонентів. Та все ж був досягнутий настільки значний прогрес, що можна говорити про те що термодинаміка є тепер важливим інструментальним засобом петрології магм.
Так само, як і у випадку взаємодії твердих силікатних тіл і розчинів електроліту, застосування термодинаміки до силікатних рідин вимагає певного рівня розуміння взаємодій що відбуваються на атомарному рівні. Тому, перед освоєнням термодинамічного підходу, варто коротко розглянути будову розплаву.
Наявна інформація щодо структури силікатних розплавів на жаль дуже обмежена й приблизна і в кращому випадку носить напівкількісний характер. Відомо, що міжатомні відстані і координаційні числа в розплаві дещо менші, ніж в кристалічній фазі того ж складу.
Більша (хоча й не вся) частка наших знань щодо структури розплавів, грунтується на вивченні скла. Звичайно, термодинамічні характеристики силікатних розплавів та їх скла відрізняються, однак дослідженнями підтверджена структурна подібність скла і рідин. Спектральні дослідження скла, яке, з певним наближенням, можна розглядати як переохолоджену рідину, свідчать що структури силікатних рідин подібні до структур відповідних твердих силікатних тіл. Фактично
129

130
головною відмінністю між силікатними розплавами і твердими тілами є відсутність впорядкування у великих фрагментах в першому випадку, в малих же фрагментах впорядкування дуже подібне. Ще в 1932 році [Zachariason W.H. The atomic arrangement in glasses. J.Am.Chem.Soc. 54, 3841-3851, 1932] було встановлено що збагачене кремнеземом скло має квазікристалічну структуру з кременекисневих ланок.
Як і в силікатних мінералах, первинним структурним елементом силікатних розплавів є кременекисневий тетраедр (див. рис. 8.4), який складається з атома кремнію, що оточений чотирма атомами кисню. Як і в силікатних мінералах, тетраедри можуть бути поєднані спільними атомами кисню, які називають містковими (тобто кожен атом кисню пов’язаний з 2 атомами кремнію і формує місткові з’єднання). Відповідно, атоми кисню, що не є спільними називаються немістковими (рис. 8.4a). Роз’єднані тетраедри кремнезему, в яких відсутні кисневі місткові з’єднання, називаються мономірами, - SiO44- (рис. 8.4b). Два тетраедри, які поєднуються одним атомом кисню називаються димірами і відповідають формулі Si2O76-. Тетраедри також можуть
бути поєднані двома атомами двома кисню і формують безкінечні ланцюги (триміри, чотириміри тощо). Хімічна формула таких ланцюгових чи розгалужених полімерів - SixO3x+12(x+1)-. В деяких
силікатах (наприклад в кварці і польовому шпаті) тетраедри повністю поєднані в каркасну структуру, і всі атоми кисню є містковими (спільними). Всі ці структури (тетраедри, ланцюги, розгалужені ланцюги, плоскі сітки, неправильні тривимірні утворення)
можуть бути присутніми в силікатному склі.
Рис.8.4. - Деякі можливі полімерні блоки силікатних розплавів. (Або) Рис.8.4. - Силікатні структури. (а): невеликі фрагменти структур силікатного розплаву подібні до структури твердих тіл. Окремі тетраедри можуть бути поєднані містковими атомами кисню, кожен з яких з’єднаний з 2 атомами кремнію. (b): найдрібнішими структурними одиницями розплавів є: мономіри (місткові з’єднання відсутні) та диміри (містковим є лише 1 атом кисню з 4).
Міра з’єднання тетраедрів кремнезему, або ступінь полімеризації, впливає на хімічні і фізичні властивості силікатних розплавів.
Одним з відображень високого ступеню полімеризації є в’язкість розплаву. Найвища ступінь полімеризації властива кремнезему, в розплаві якого більшість атомів кисню є містковими, а низька
електропровідність свідчить про домінування ковалентних зв’язків. Домішка невеликих кількостей окислів металів звичайно викликає зниження в’язкості (наприклад, домішка 2,5 мол. % К2О при температурі 1700 викликає зменшення в’язкості з 2000000 до 200 пуаз - динамічна в’язкість 1П=0,1 Па*с) та різке збільшення провідності, що свідчить про зменшення ступеню полімеризації за рахунок розриву частини місткових зв’язків та про те, що зв’язок між металом та киснем є переважно іонним (дрібні іони рухаються швидше, ніж крупні). Зміна в’язкості залежить не тільки від концентрації домішок, але й від виду іонів металів:
наприклад, Са2+ викликає значно сильнішу деполімеризацію, ніж Со2+.
Рис. 8.5. Структура чистого скла кремнезему (a) і скла
кремнезему збагаченого іонними домішками (b). Сіткоутворювачі: Si4+, Al3+, P3+, інколи Fe3+, Ti4+, B3+.
Сіткомодифікатори: Ca2+, Mg2+, K+, Na+, Fe2+, H+, інколи
Ti4+, Al3+.
Зменшення ж в’язкості призводить до зростання швидкості ряду
важливих процесів, таких як ріст мінералів та осадження кристалів.
****
Полімерні моделі.
130