

22
3.3. Використання автоматичної перевірки та аналізу побудови хімічної структури
Досить часто в практиці хіміка виникає необхідність перетворити структурну формулу сполуки у звичайну строкову.
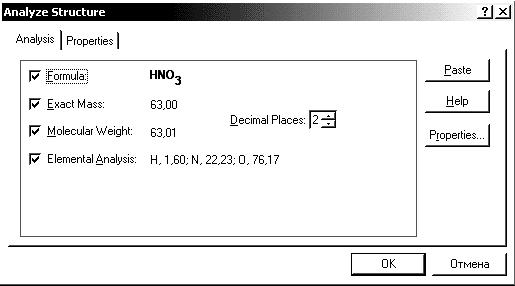
Рис. 3.1. Приклад використання функції Analyze structure.
Це досить легко зробити за допомогою пункту меню Structure>Analyze structure. Після вибору цієї команди з’явиться вікно, що відображає не тільки формулу сполуки, але і молекулярну вагу та процентну частку атомів кожного елементу, що входять в молекулу (див. рис. 3.2).
Рис. 3.2 Вікно інформації Analyze structure.
Для вставки інформації з вікна Analyze structure в формулу виберіть кнопку Past. Результат див. на рис. 3.3.
23
OH
O- N+ O
HNO3
Exact Mass: 63,00
Mol. Wt.: 63,01
H, 1,60; N, 22,23; O, 76,17
Рис. 3.3 Результат роботи команди Analyze structure.
Програма також перевіряє наявність помилок (не вірна валентність, некоректно вибрано тип зв’язку) у створених формулах. Для використання цієї функції треба вибрати пункт меню Structure>Check structure. Програма знайде помилку, з'явиться діалогове вікно, в якому буде вказано атом з некоректним зв’язком чи валентністю. Якщо програма помилок не знайде не яких повідомлень не буде. Програма автоматично перевіряє коректність написання структури весь час роботи, іноді це заважає. Для відключення цієї функції виберіть пункт меню: Tools>Show Chemical Warnings.
Програма має достатньо велику базу даних найбільш сполук, що часто використовуються. За допомогою засобу Convert Name To Structure - конвертація імені в структурну формулу можливи досить швидко створювати формули сполук, що присутні в базі програми. Досить ввести назву сполуки та вибрати пункт меню Structure>Convert Name To Structure. Якщо сполука присутня у базі програма створить її структурну формулу.
Засобами ChemDraw можливо вирішити зворотну задачу знайти коректну назву для сполуки. Досить виділити формулу сполуки та вибрати пункт меню Structure>Convert Structure To Name. Під формулою з’явиться назва сполуки, якщо вона присутня у базі даних програми.
24
Лекція 4.
Створення тривимірних моделей молекул за допомогою спеціального програмного забезпечення
Певну складність для студентів, які вивчають біоорганічну хімію, становить формування стереохімічних уявлень про молекули. Такого роду знання особливо важливі, враховуючи існування тісного взаємозв'язку між просторовою будовою молекул і біологічною активністю сполук.
Chem3D - программа, разработанная, для моделирования химических соединений в трехмерном виде. Она объединяет возможности визуализации, анализа, и вычислительные инструменты с удобным в работе графическим пользовательским интерфейсом. В Chem3D используются инструменты, основанные на молекулярной механике созданные для оптимизации моделей, конформационный поиск, молекулярную динамику.
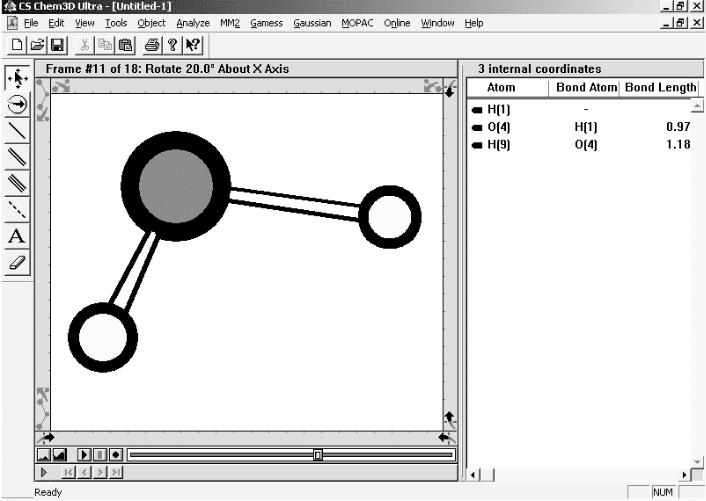
Как и любая программа Chem3D состоит из заголовка окна с тремя управляющими кнопками, главного меню, панелей инструментов и рабочей области.
Рис. 1. Окно программы Chem3D.
25
Главное меню программы состоит из следующих пунктов: File, Edit, View, Tools, Object, Analyz, MM2, Gammes, Gaussian, MOPAC, Online, Window, Help.
File – в этом пункте меню собраны все команды относящиеся к работе с файлами: Пункт New Model (Новая модель) открывает новое окно пользователя. Отметим,
что Chem3D Ultra 6.0 работает в многооконном режиме.
Пункт Templates (Заготовки) предлагает открыть новое окно с уже заданными настройками (например, цвет фона или способ отображения молекул) или с заготовками моделей молекул (например, кристалл NaCl рис. 2).
Пункты Open/Close Window/Save/Save As.. (Открыть/Закрыть окно/Сохранить/Сохранить как...) представляют собой стандартные функции открытия, закрытия и сохранения файлов.
Пункт Revert to Saved (Вер1гуться к сохраненному) убирает все изменения после последнего сохранения.
Пункт Set Default Settings (По умолчанию) использует установки данной модели для новой модели.
Пункты Print/Print Setup (Печать/Настройка) открывает стандартное диалоговое окно для настройки и последующей распечатки файла.
Edit – Пункт меню Edit (Правка) также предоставляет обычный набор функций копирования, вставки, отмены последнего действия и выбора.
Пункт Undo/Redo (Отменить/Вернуть) отменяет или, наоборот, возвращает изменения последнего действия.
Пункт Cut (Вырезать) вырезает выбранный элемент молекулы/текст и копирует его в буфер обмена.
Пункт Copy (Копировать) копирует выбранный элемент молекулы/текст в буфер обмена.
Пункт Paste (Вставить) вставляет выбранный элемент молекулы/текст из буфера обмена.
Пункт Clear (Очистить) удаляет выбранный элемент/текст.
Пункт Copy As (Копировать как...) позволяет копировать выделенное как ChemDraw — структуру или растр (Bitmap).
Пункт Select All (Выбрать все) выбирает все элементы.
Пункт Select Atoms (Выбрать атомы) позволяет выбирать атомы по определенному признаку: Reverse (Выбрать невы-бранные, т. е. обратить выделение), Select С (Выбрать все атомы одного элемента), Select Adjacent (Выбрать атомы, смежные к данному), Select Fragment (Выбрать все атомы данного фрагмента).
Пункты Clear Frames/Molecular Surfaces/Calculations (Очистить) позволяет очистить кадры/молекулярные поверхности/расчеты.
View – Пункт меню View (Вид) предоставляет широкий ассортимент настроек вида молекл/связей/панелей и т. д. Он состоит из трех разделов.
Первый раздел включает три пункта: галочку Toolbar (Главная панель), включающую отображение главной панели; галочку
26
Tools Palette (Контрольная панель), отображающую видимость контрольной панели, и пункт Settings (Настройки), выводящий диалоговое окно настройки вида молекул. Его назначение мы рассмотрим позднее.
Второй раздел объединяет пункты, с помощью которых можно визуализировать различные расчетные характеристики молекулярной модели. Подробно соответствующие расчетные методы компьютерной химии будут рассмотрены в последующих главах. Здесь же мы лишь опишем назначение отдельных пунктов.
Пункт Solvent Accessible Surface (Поверхность, доступная растворителю) отображает модель молекулы в виде поверхности, доступной молекулам растворителя. Поверхность изображается на основании расчета методом Конопли площади, заметаемой центром пробной сферы (моделирующей рас-юоритель) при ее мысленном прокатывании по поверхности исследуемой молекулы. При этом атомы в модели молекулы представляются в виде сфер с радиусами, равными вандерваальсовским радиусам соответствующих атомов. Это окно включает следующий набор настроек:
Solvent Radius (Радиус растворителя) может быть установлен значениями от 0,1 до 10 А движением бегунка. По умолчанию радиус растворителя установлен равным 1.4 А, соответствующим воде.
Map Property (Свойста поверхности) предлагает настройки цвета отображения поверхности.
Resolution (Разрешение) устанавливает качество отображаемой поверхности от грубого до тонкого. Как видно, при низком разрешении поверхность, доступная растворителю, становится почти эллипсоидальной, тогда как при высоком разрешении она почти повторяет исходную модель молекулы.
Пункт Connolly Molecular Surface (Молекулярная поверхность Конолли) предлагает аналогичные настройки. В данном случае для построения поверхности используется другой алгоритм Конолли, в котором поверхность определяется контактом с молекулой растворителя — сферой с заданной величиной ван-дер-ваальсовского радиуса. Для сравнения на рис. 1.31 приведена поверхность Конолли с разрешением 10 той же модели, что и на рис. 1.30. Видно, что алгоритм Конолли дает более детальную оценку поверхности.
Пункт Total Charge Density (Общая плотность заряда) позволяет визуализировать пространственную плотность распределения заряда — электронную плотность в пространстве, окружающем ядро молекулы, или вероятность нахождения электронов в окружении молекулы.
Окно настроек позволяет задать метод расчета (Calculation), тип поверхности (Surface Туре), ее цвет и т. д.
Пункт Total Spin Density (Спиновая плотность) описывает пространственное распределение разницы в плотностях между Т-спиновыми и 1-спиновыми электронами в данной точке.
Пункт Molecular Electrostatic Potential (Молекулярный элект-I юстатический потенциал) позволяет оценить распределение молекулярного электростатического потенциала. Последний характеризует притяжение или отталкивание между молекулой и протоном в данной точке. Притяжение определяется отрицательными значениями потенциала, а отталкивание — положительными.
27
Пункт Molecular Orbitalis (Молекулярные орбитали} позволяет показать поверхности, визуально отображающие различные стабильные электронные распределения в молекуле. Расчет их может быть проведен квантово-химическими методами.
Пункт Partial Charges (Частичные заряды) используется для отображения частичных зарядов на атомах, вычисленных на основании квантово-химического расчета.
Впоследнем разделе пункта меню View приведены ссылки на таблицы с параметрами, используемыми в расчетах. Например, таблица ММ2 Atom Type Parameters содержит поля с параметрами атомов, используемыми при расчете энергии молекулы методом молекулярной механики.
Tools – Пункт меню Tools (Инструменты) предлагает инструменты отображения различных панелей, меню, функции масштабирования и отражения молекулы.
Впервом разделе сосредоточены пункты настройки рабочего окна программы. Галочка Show Status Bar включает отображение строки состояния.
Галочка Show Rotation Bars включает отображение средств вращения молекулы в
окне визуализации.
Галочка Show Movie Controller включает отображение контроллера анимации. Галочка Show Messages включает отображения поля с сообщениями.
Галочки Show Model Table включает отображения полей с различными данными (измерения, группы, координаты).
Галочка Show H's and Lp's включает отображение атомов водорода и неподеленных электронных пар в модели молекулы.
В следующих разделах сосредоточены пункты, позволяющие управлять отображением модели молекулы в окне редактора модели.
Функции Magnify (Увеличить) и Reduce (Уменьшить) соответственно изменяют масштаб молекулы в окне визуализации.
и М в нижней левой части окна модели, а также горячие клавиши F7 и F8 соответственно.
Функция Rectify (Исправить) проверяет наличие в молекуле связей, согласно химическим правилам. Например, если четырехвалентный углерод содержит только две связи, будут добавлены два атома водорода. По умолчанию это делается автоматически.
Функция Clean Up Structure (Очистить) проверяет соответствие в модели молекулы длин связей и валентных углов принятым в химии стандартным значениям. Если соответствие отсутствует (например, когда молекула рисовалась вручную), то делаются необходимые исправления в ее структуре.
Функция Dock (Стыковать) ориентирует один фрагмент относительно другого. Функция Overlay (Покрыть) покрывает один фрагмент другим с помощью атомных
пар.
Пункт Reflect (Отразить) позволяет отразить молекулу относительно заданных плоскостей или инвертировать ее относительно начала координат.
Пункт Fit Model to Window (Вписать модели в окно) изменяет масштаб молекулы таким образом, чтобы она была полностью видна в окне визуализации.
Пункт Fit Selection to Window (Вписать выделение в окно) изменяет масштаб выделенной части модели (Selection) таким образом, чтобы она была полностью видна в окне визуализации.
28
Пункт Fit All Frames to Window (Вписать все кадры в окно) изменяет масштаб всех кадров анимации таким образом, чтобы они были полностью видны в окне визуализации.
Object – Пункт меню Object (Объект) (рис. 1.36) предлагает инструменты настройки отображения различных параметров молекулы, а также измерения длин связей, межатомных расстояний и углов.
Функция Move to Center (Двигать в центр) перемещает молекулу или выделение в центр окна визуализации.
Функция Move To (Двигать в...) перемещает молекулу или выделение на соответствующую ось X, Y или Z.
Пункт Colorize (Изменить цвет) дает возможность изменить цвет атомов по желанию с помощью палитры.
Пункт Show Elements Symbols (Показать символы элементов) позволяет отображать символы элементов (например, С, Н, С\ и т.д.).
Пункт Show Serial Numbers (Показать порядковые номера) отображает порядковые номера атомов в модели молекулы, установленные программой.
Пункт Show Solid Spheres (Показать твердые сферы) включает отображение атомов в виде сплошных сфер.
Пункт Show Dot Surfaces (Показать точечные поверхности) включает отображение атомов в виде сфер с вандерваальсовскими радиусами посредством точек.
Функции Set Distance (Определить расстояние), Set Bond Length (Определить длину связи), Set Bond Angle (Определить угол) и Set Dihedral Angle (Определить двугранный угол) позволяют измерить и при необходимости изменить соответствующие расстояния и углы в модели молекулы. Для того чтобы моспользоваться ими, необходимо иыделить с помощью мыши атомы, ()бразующие нужную связь или формирующие нужный угол, и выбрать соответствующую функцию. Результат измерения будет отображен в окис Measurements (Измерения) в поле Actual (Фактическое). Кроме того, в иоле Optimal будет отображено значение данной величины, принятое в качестве стандартного для выбранной связи или угла. При необходимости изменить выделенное расстояние между атомами или угол, следует набрать с клавиатуры требуемое значение в поле Actual и нажать клавишу ввода или щелкнуть мышью в окне редактора.
Функция Break Bond (Разорвать связь) разрывает выделенную связь.
Пункт Set Bond Order (Установить порядок связи) изменяет порядок связи на одинарный/двойной/тройной.
Функция Bond Proximate (Ближайшая связь) создает химическую связь между двумя ближайшими атомами. Ближайшими в программе считаются два атома, расстояние между которыми меньше, чем принятое стандартное значение для длины связи плюс некоторое значение, величину которого (в процентах от длины связи) можно изменить в настройках пункта меню View\Settings\Building.
Пункт Add Centroid (Добавить центровой) создает атом-пустышку в центре выделенной молекулы. Эта функция удобна при моделировании координационных соединений.
Функция Invert (Инвертировать) выполняет операцию пространственной симметрии в выбранной группе хиральных атомов.
29
Пункт Set 2 Matrix (Установить Z-матрицу) позиционирует выбранный атом в Z- матрице молекулы. Относительное положение каждого атома в молекуле определяется набором переменных, образующих так называемую Z-матрицу молекулы. С помощью данного пункта можно выбрать атом, который в системе отсчета Z-матрицы будет установлен в качестве начального.
Функции Hide/Show (Скрыть/Отобразить) позволяет скрыть (видимость) выбранных атомов или отобразить их в окне визуализации.
Пункт Define Group (Определить группу) позволяет сгруппировать выбранные атомы под определенным именем.
Analyz – Пункт меню Analyze (Анализировать) (рис. 1.38) предлагает инструменты анализа молекул и расчета их свойств.
Пункты Spin About X/Y/Z/Selected Axis (Вращать) включают режим вращения молекулы относительно оси X/Y/Z или выбранной оси. С помощью кнопок меню анимации возможно записать анимацию, а затем воспроизвести ее.
Пункт Spin Torsional Angles (Вращать двугранные утлы) вызывает операцию вращения определенных групп атомов относительно двугранного угла.
Пункт Show Measurements (Показать измерения) открывает окно Measurements, в котором отображаются значения длин связей и валентных и двугранных углов для всех атомов молекулы. gpin About X Axis
Пункт Extended Huckel Surfaces (Поверхности расширенного метода Хюккеля) позволяет рассчитать молекулярную поверхность расширенным методом Хюккеля.
Метод Хюккеля — это один из простеиших полуэмпирических квантово-химиче- ских методов расчета. Вычисления с его помощью осуществляются наиболее быстро. Следует заметить, однако, что при его использовании не следует забывать о Compute Properties. После проведения расчета выберите какую-нибудь молекулярную поверхность (молекулярную орбиталь) в меню View (Вид) и отобразите ее кнопкой Show Surface (Показать поверхность). Аналогичным образом можно отобразить распределение частичных зарядов в молекуле.
Пункт Deviation from Plane (Отклонение от плоскости) показывает отклонения выбранных атомов от нормальной плоскости.
Пункт Compute Properties (Расчет свойств) вызывает одноименное окно (рис. 1.39), позволяющее выводить результаты расчета выбранного свойства или группы свойств молекулы или соотвествующего вещества. Данный пункт объединяет различные расчетные методы, представленные в пакете Chem3D. 11одробнее об этом рассказывается в главах, посвященных соответствующим методам компьютерной химии.
Пункты меню ММ2 (Молекулярная механика), Gamess, Gaussian и МОРАС предлагают методы расчета потенциаль-I юй энергии молекул и прочих характеристик эмпирическими, полуэмпирическими и неэмпирическими квантово-механичес-кими методами.
MM2, Gammes, Gaussian, MOPAC – Пункты меню ММ2 (Молекулярная механика), Gamess, Gaussian и МОРАС предлагают методы расчета потенциаль-I юй энергии молекул и прочих характеристик эмпирическими, полуэмпирическими и неэмпирическими квантово-механичес-кими методами.

30
Мы не будем подробно останавливаться на каждом пункте этих меню, рассмотрим лишь основные общие черты.
Пункт Run MM2 Job... (Запустить ММ2 задачу) открывает диалоговое окно для выбора файла описания задания ММ2 с расширением *.jdf или *.fdt.
Пункт Minimize Energy... (Минимизировать энергию) запускает процесс минимизации потенциальной энергии молекулы.
Пункт Molecular Dynamics... (Молекулярная динамика) активизирует молекулярнодинамическии эксперимент с возможностью задания температуры, числа итераций и других параметров.
Пункт Compute Properties... (Расчет свойств) открывает окно для выбора типа рас-
чета.
Пункт Show Used Parameters (Показать использовавшиеся параметры) отображает параметры ММ 2 в окне Messages (Сообщения).
Online – получение информации с сайта производителя программы (запрос в закрытые базы данных).
Window – здесь собраны команды для изменения расположения открытых окон. Help – получение справочной информации по работе с программой (англ. яз.).
Контрольная панель располагается в левой части экрана имеет небольшой набор
необходимых средств: |
|
- создание нового документа; |
(1) |
- вызов диалогового окна открытия документа; |
(2) |
- сохранение документа; |
(3) |
- вырезать объект в буфер обмена; |
(4) |
- скопировать объект в буфер обмена; |
(5) |
- вставить объект в буфер обмена; |
(6) |
- отмена последней выполненной операции; |
(7) |
- выделение объекта; |
(8) |
- вращение объекта; |
(9) |
- создание связи; |
(10) |
- создание двойной связи; |
(11) |
- создание тройной связи; |
(12) |
- создание атома молекулы; |
(13) |
- удаление связи или атома из молкулы; |
(14) |
С помощью инструмента 13, если щелкнуть по пустому месту в окне визуализации, то будет дана возможность создать новый фрагмент, просто записав формулу, например С2Н5ОН.
Існує кілька способів створення тривимірної моделі молекул: а) написання у ChemDraw структурної формули, а потім копіювання її в вікно
31
Chem3D. При вставці генерується тривимірна модель, при цьому різним довжинам зв'язків і валентних кутах присвоюються відповідні стандартні значення; б) трансформація молекулярної формули, записаної за певним алгоритмом у поле введення тексту вікна Chem3D, в просторову модель одного з ізомерів. Можливо, що створена модель зажадає подальшого редагування; в) безпосереднє редагування тривимірної моделі будь-якої складної конфігурації у вікні Chem3D. Цей спосіб є основним. У процесі редагування можна змінювати розташування моделі або окремих її частин в просторі для кращої наочності. Програма Chem3D допускає різні способи візуалізації тривимірної моделі молекули: напівсферична Стюарта-Бріглеба, молекулярна поверхня Коноллі (визначається контактними точками з молекуламирозчинника), шаростержневие, стрижнева, ін
Использование двумерной модели, созданной в одном из простых химических редакторов. Двумерная модель в виде структурной формулы соответствующего соединения может быть создана в приложении CS ChemDraw или в другом «химическом редакторе», например ISIS/Draw, а затем перенесена в окно Chem3D через буфер обмена Windows при помощи операций «копировать» и «вставить». При вставке в окно Chem3D структурная формула автоматически преобразуется в трехмерную модель, при этом соответствующим длинам валентных связей и углов между атомами присваиваются стандартные для данных элементов значения. Для тех редакторов, с которыми Chem3D не поддерживает интерфейса через буфер обмена Windows, импорт модели может быть осуществлен через файл одного из стандартных химических форматов (файлы с расширением .ale, .mol, .msm, .mop и многие другие).
Написание брутто-формулы соединения в рабочем поле окна. После нажатия кнопки с буквой «А» на панели инструментов курсор приобретает вид перекрестья с буквой «А» в центре. Если щелкнуть мышью в любом месте рабочего окна, появляется поле ввода текста, в котором можно заглавными латинскими буквами набрать бруттоформулу соответствующего соединения, например C2H5OH. После нажатия клавиши ввода в рабочем окне будет создана трехмерная модель соответствующего соединения. Следует иметь в виду, что одной и той же брутто-формуле могут отвечать различные структурные изомеры, поэтому возможно, что созданная модель потребует затем дальнейшего редактирования.
Непосредственное редактирование с использованием кнопок на панели инструментов. Этот способ является основным, поскольку позволяет создать трехмерную модель любой сложной конфигурации. При нажатии одной из кнопок на панели инструментов, соответствующей одинарной (наклонная черта), двойной (две наклонных черты) или тройной (три наклонных черты) связи, курсор в рабочем поле окна принимает вид перекрестья. Для того чтобы получить трехмерную модель молекулы из двух углеродных атомов, достаточно провести черту при помощи левой клавиши мыши в рабочем поле окна. Нужное количество атомов водорода, соответствующее выбранному типу связи, будет добавлено автоматически. Дальнейшее редактирование может проводиться добавлением новых связей взамен выбранных атомов водорода, изменением геометрии модели, путем перетаскивания мышью выбранных атомов, созданием циклов, путем переноса и последующего наложения нужных атомов углерода, образующих цикл, изменением типа связи. Для того чтобы изменить тип связи, нужно щелкнуть на кнопке на панели инструментов, соответствующей нужному типу связи, и с нажатой клавишей
32
мыши соединить чертой два выбранных атома углерода. Для создания модели с гетероатомами, например кислорода или азота, нужно выбрать на панели инструментов кнопку текстового ввода (кнопка с буквой «А»), выделить щелчком мыши нужный атом (водорода или углерода) и набрать в текстовом поле символ необходимого атома или брутто-формулу заместителя. В процессе редактирования возможно вращение созданной модели или отдельных ее частей в пространстве, изменение размеров отображаемой области, отражение и перемещение. Вращение модели в двух перпендикулярных плоскостях производится при помощи полос прокрутки в нижней и правой частях рабочей области окна. При помещении курсора в область полос прокрутки он приобретает вид ладони. Перемещая в таком состоянии курсор с нажатой левой клавишей мыши вдоль полосы прокрутки, можно вращать модель вокруг горизонтальной и вертикальной оси (правая вертикальная и нижняя полосы прокрутки соответственно). Если необходимо вращать отдельные элементы модели вокруг выбранной связи, нужно выделить нужную связь, выбрав щелчком мыши один или более нужных атомов. При необходимости выделить несколько атомов нужно щелкать на них, удерживая нажатой клавишу Shift, или обвести атомы рамкой с нажатой клавишей мыши. Вращение группы атомов вокруг выделенной связи производится перемещением левой вертикальной полосы прокрутки Выбор клавиши Jsa на панели инструментов позволяет одновременно вращать модель в двух плоскостях в произвольном направлении. Изменение размеров отображаемой области может осуществляться нажатием кнопок в нижней части рабочего окна (левая кнопка соответст-пует уменьшению размеров модели, правая — увеличению) или ныбором пунктов меню Tools/Reduce (уменьшить, функциоальная клавиша F8) и Tools/Magnify (увеличить, функциональая клавиша F7). Чтобы переместить в плоскости экрана группу атомов, нужно выделить их и, установив указатель курсора на один из выделенных атомов, перемещать по экрану с нажатой левой клавишей мыши.
33
Лекція 5.
Хранение и поиск информации в области текстильной химии и химической технологии с помощью программы ChemFinder
Химическая информация разбросана по большому числу источников, включая научные журналы, патенты, доклады, диссертации, материалы конференций, книги. Объем химической информации растет чрезвычайно быстро.
Современный химик, если он действительно не хочет отстать то развития своей дисциплины, должен уделять химической информации много времени.
Число разнообразных публикаций сейчас настолько велико, что помощь компьютера становится совершенно необходимой. Если суммировать ежегодно появляющиеся и имеющие отношение к химии статьи, патенты, доклады, диссертации, то окажется, что за одну минуту на свет появляется несколько статей. Так например самое крупное периодическое издание «Chemical Abstracts» публикует в год около 500000 статей. К этому необходимо добавить все публикации, имеющие отношение к химии, но не представленные в этом издании.
Увеличение количества доступной информации требует трудным получение за разумное время полного представления о конкретной задаче, если не использовать возможности компьютерной техники. Именно по этому непосредственный доступ к информации становится из года в год все более необходимым во всех областях химии.
С развитие Интернет практически каждый может получить доступ к открытым (бесплатным) или закрытым (платным) базам знаний. Но получение информации это только первый этап работы с ней. Далее ее нужно сохранить и систематизировать в таком виде.
Задача хранения и структурирования больших объемов данных наиболее эффективно решается с помощью баз данных.
В настоящий момент не существует однозначного поределения термина «База данных», наиболее распространенные поределения:
База данных — это информационная модель, позволяющая упорядоченно хранить данные о группе объектов, обладающих одинаковым набором свойств.
Базами данных являются, например, различные справочники, энциклопедии и т. п. Базой данных является представленная в объективной форме совокупность самостоятельных материалов (статей, расчетов, нормативных актов, судебных решений и иных подобных материалов), систематизированных таким образом, чтобы эти материалы
могли быть найдены и обработаны с помощью электронной вычислительной машины.
Информация в базах данных хранится в упорядоченном виде. Так, в записной книжке все записи упорядочены по алфавиту, а в библиотечном каталоге либо по алфавиту (алфавитный каталог), либо в соответствии с областью знания (предметный каталог).
Существует несколько различных типов баз данных: табличные, иерархические и сетевые.
Табличные базы данных являются наиболее распространенными. Табличная база данных содержит перечень объектов одного типа, т. е. объектов с одинаковым набором свойств. Такую базу данных удобно представлять в виде двумерной таблицы.

34
Столбцы такой таблицы называют полями; каждое поле характеризуется своим именем (названием соответствующего свойства) и типом данных, отражающих значения данного свойства. Поля Название и Тип процессора — текстовые, а Оперативная память
— числовое. При этом каждое поле обладает определенным набором свойств (размер, формат и др.). Так, для поля Оперативная память задан формат данных целое число.
Поле базы данных — это столбец таблицы, включающий в себя значения определенного свойства.
Строки таблицы являются записями об объекте; эти записи разбиты на поля столбцами таблицы. Запись базы данных — это строка таблицы, которая содержит набор значений различных свойств объекта.
В каждой таблице должно быть, по крайней мере, одно ключевое поле, содержимое которого уникально для любой записи в этой таблице. Значения ключевого поля однозначно определяют каждую запись в таблице.
Для хранения боьших объемов информации химической тематики разработаны специальные приложения. К их числу относиться программа ChemFinder, входящая в пакет ChemOffice.
Программа позволяет создавать и просматривать базы данных и эффективно проводить в них поиск нужной информации. Отличительной особенность является возможность удобной работы с химическими структурными формулами. Это позволяет не только хранить в базе текстовую информацию но и изображения формул.
Как и любая программа ChemFinder состоит из заголовка окна с тремя управляющими кнопками, главного меню, панелей инструментов и рабочей области.
Рис. 1. Вікно програми ChemFinder.
Главное меню программы состоит из следующих пунктов: File, Edit, View, Text, Search, Record, Script, Online, Window, Help.
35
File – в этом пункте меню собраны все команды относящиеся к работе с файлами: New – создает новый файл; Open – открывает существующий файл, Save – сохраня-
ет изменения слеланные в файле, а если файл еще не сохранен вызывает диалоговое окно сохранения файла; Close – закрывает текущий файл; Database – вызывает диалоговое окно мэнэджера баз данных, с помощью которого можно создавать, удалять, изменять и редактировать базы данных; Import – импортирует готовую базу данных в активный файл; Export – экспортирует из файла базу данных; Page Setup – вызывает диалоговое окно позволяющее изменять параметры страницы при печати базы данных; Preferences – общие настройки базы данных; Exit – выход из ChemFinder.
Edit – в этом пункте меню собраны все команды относящиеся редактированию содержимого файла и работы с буфером обмена: Undo – отмена последней операции; Redo – возврат к последней выполненной операции; Cut – вырезает объект в буфер обмена; Copy – копирует объект в буфер обмена; Paste – вставляет объект в базу.
View – в этом пункте меню собраны все команды относящиеся к изменению отображения данных открытой базы: Data Table – просмотр базы данных в виде таблицы; Periodic Table – вызов окна с периодической таблицей Менделеева; Greed – включает/выключает отображение сетки в форме; Switch view – переключатель вида отображения данных таблица/форма.
Text – в этом пункте меню собраны все команды относящиеся к работе с текстом: Format – изменяет формат текста; Style – изменяет стиль текста – надстрочный/подстрочный/жирный/курсив/подчеркнутый; Size – изменяет размер текста; Flush Left, Flush right, Flush center – соответственно выравнивает текст по левому полю, правому и по центру.
Search – в этом пункте меню собраны все команды относящиеся к поиску нужной информации в открытой базе данных: Enter Query – переключение в режим создания запроса, при этом пользователь должен ввести в соответствующее поле либо часть имени, либо часть формулы, либо молекулярный вес соединения; Find – выполняет поиск по параметрам заданным в запросе; Abort Query – останавливает действие запроса; Preferences – вызывает диалоговое окно настройки поиска.
Record – в этом пункте меню собраны все команды относящиеся к работе с записями открытой базы данных; First Record – переход к первой записи базы данных; Previous Record - переход к предыдущей записи базы данных; Next Record - переход к следующей записи базы данных; Last Record - переход к последней записи базы данных; Go to Record - переход к записи базы данных под номером…; Add New Record – добавить новую запись в базу; Delete Record – удалить текущую запись из базы.
Script – Command line – позволяет пользователь запускать часто повторяющиеся действия одной командой.
Online – получение информации с сайта производителя программы (запрос в закрытые базы данных).
Window – здесь собраны команды для изменения расположения открытых окон. Help – получение справочной информации по работе с программой (англ. яз.).
- создание нового документа; |
(1) |
- вызов диалогового окна открытия документа; |
(2) |

36 |
|
- сохранение документа; |
(3) |
- вырезать объект в буфер обмена; |
(4) |
- скопировать объект в буфер обмена; |
(5) |
- вставить объект в буфер обмена; |
(6) |
- отмена последней выполненной операции; |
(7) |
- переход к первой записи; |
(8) |
- переход к предыдущей записи; |
(9) |
- переход к записи базы данных под номером…; |
(10) |
- переход к следующей записи; |
(11) |
- переход к последней записи; |
(12) |
- добавить новую запись; |
(13) |
- переключение в режим создания запроса; |
(14) |
- поиск по запросу в базе |
(15) |
- выделение объекта; |
(16) |
- вставка поля данных; |
(17) |
- вставка поля с фреймом; |
(18) |
- вставка надписи; |
(19) |
- вставка рисунка; |
(20) |
- вставка кнопки; |
(21) |
- вставка подформы; |
(22) |
- включение/отключения отображения сетки. |
(23) |
Для создания собственной базы химических соединений нужно выполнить следующие действия:
1.Создать новый файл File > New, при этом создается новая форма.
2.Вызвать контекстное меню, нажав на поле правой кнопкой мыши выбрать пункт Data source. В появившемся диалоговом окне выбрать кнопку Create Database. Назвать базу, например «chem.mdb» (редактор автоматически добавляет расширение файла .mdb, поэтому его не нужно указывать в поле названия файла). Программа автоматически создаст в базе 4 типа данных: для структурной формулы, молекулярной массы, формулы и порядкового номера записи в базе данных. Для того чтобы в базе хранилось и название соединения нужно перейти во вкладку Field нажать на кнопку Create field в поле Name ввести название поля Compounds name в списке Type выбрать Data Box.
3.С помощью инструмента 18 вставка поля с фреймом создать 5 полей. Вызвать контекстное меню для каждого, нажав на поле правой кнопкой мыши, выбрать пункт Properties, назвать поля: Структурная формула, Название соединения, Формула, Молекулярная масса, Порядковый номер.
4.Сохраните базу, назвав ее соответственно содержимому. Например, «Препараты для заключительной отделки.cfw» (редактор автоматически добавляет расширение файла .cfw, поэтому его не нужно указывать в поле названия файла).
37
5.Теперь база готова для работы. Чтобы ввести в нее новую запись нужно в меню Record выбрать пункт Add New Record – добавить новую запись в базу.
6.Вызвать контекстное меню, нажав на поле правой кнопкой мыши, выбрать пункт Edit structure. Автоматически запуститься ChemDraw. Создайте структурную формулу соединения. Закройте ChemDraw. В поле появится структурная формула, которую вы только что создали.
7.В поле название соединения введите название. Поля «Молекулярная масса» и «Порядковый номер» заполняются программой автоматически.
8.Чтобы добавить следующую запись снова выполните действия из пунктов 5-7.
9.При завершении работы с базой сохраните ее, выбрав меню File > Save.
ChemFinder позволяет так же хранить и текстовую информацию, например архив статей. Затрачивая некоторое время на ввод данных в такую базу, пользователь может затем экономить много времени при поиске нужной информации. В зависимости от размера базы скорость поиска может возрасти в 10-20 раз по сравнению с поиском информации в печатном виде.