

Gale Encyclopedia of Genetic Disorder / Gale Encyclopedia of Genetic Disorders, Two Volume Set - Volume 2 - M-Z - I
.pdfRetinoblastoma
Child with a large tumor proturuding from the right eye socket. (Custom Medical Stock Photo, Inc.)
retinoblastoma, this growth and division is uncontrolled, leading to cancer of one or both eyes.
In 40% of children who present with retinoblastoma, there will be an abnormality of chromosome 13 in every cell in that child’s body, including the affected eye(s). In 60% of the children who have retinoblastoma, the abnormal chromosome will only be found in the eye.
As stated earlier, retinoblastoma can occur spontaneously (nonhereditary) or be seen in families (hereditary). If neither parent had retinoblastoma, then the chances of having a child with retinoblastoma is approximately one in 20,000. However, parents with a child having the condition should have a detailed retinal exam to see if they perhaps had retinoblastoma and were not aware of it. One percent of the time, this exam will reveal that one of the parents had a limited form of retinoblastoma that was never diagnosed. In this case, a full 45% of the parent’s children will have the chance of developing retinoblastoma. In even rarer cases, the parent could have the gene for retinoblastoma without any evidence of the disease in their eyes. This is called a carrier state, and again, 45% of the parent’s children will have the chance of developing retinoblastoma. There are now genetic tests to determine if a parent is a carrier of the condition. However, the current test is only 80% accurate, and costs between $2,500.00 and $4,000.00.
In hereditary retinoblastoma, if a parent has bilateral (seen in both eyes) retinoblastoma and decides to have children, then with each pregnancy, there is a 45%
chance his or her children will develop retinoblastoma. Many of these children will also have tumors spread from their retinas into their brains at the time of birth. Other children will not develop tumors until they are two to three years of age. Most children born to a parent with bilateral retinoblastoma will also have the condition in both eyes. Fifteen percent of these children will have retinoblastoma in only one eye.
There is also hereditary retinoblastoma in which the parent has unilateral (in one eye) retinoblastoma. In this case, 7–15% of the children of these parents will develop retinoblastoma. Like the children born to a parent with bilateral retinoblastoma, most of the children (approximately 85%) born to parents with unilateral retinoblastoma will have bilateral retinoblastoma.
Demographics
Retinoblastoma is the most common type of eye cancer in children. It occurs in approximately one in 20,000 births, which means that each year about 200–300 children are affected in the United States. The incidence of retinoblastoma in other areas of the world is thought to be approximately the same. As more and more children survive this condition and grow into adulthood and have their own families, the frequency of the condition in the population will probably increase. Retinoblastoma affects children of all races and is seen equally in both boys and girls.
Signs and symptoms
Since the successful management of retinoblastoma depends on detecting it early, the recognition of the signs and symptoms of the condition is critical. This is especially true for primary care physicians, who are often the first medical personnel to see infants or children with retinoblastoma.
There are many ways that retinoblastoma can present itself in infants and children. More than half of all patients with the condition will have a white pupil reflex, called leukocoria. In healthy infants and children, their pupils will appear black, or, when photographed, red. However, patients with retinoblastoma will often have a pupil that appears gray or white.
The second most common presenting sign of retinoblastoma, occurring about 25% of the time, is a crossed eye, a medical condition referred to as strabismus. The child’s eye may appear to be looking out towards the ear, called exotropia, or inward towards the nose, called esotropia. It should be noted that three to four percent of all American children present with some form of strabismus, but not all of these children have
992 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
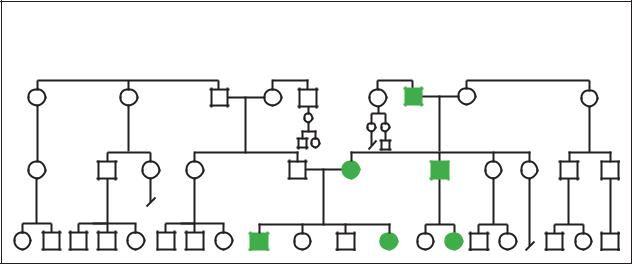
Retinoblastoma |
Retinoblastoma |
Autosomal Dominant (40%) |
|
|
(Gale Group)
retinoblastoma. However, since 25% of children with retinoblastoma have strabismus, any child with this condition should have a detailed eye exam to rule out retinoblastoma.
While leukocoria and strabismus are the two most common presenting signs of retinoblastoma, there are other ways the condition may present itself. Other symptoms may include a red, painful eye, poor vision, orbital cellultis (inflammation of the skin and tissue around the eye), and amblyopia, or “lazy eye.” Heterochromia, which is different colored irises (the colored part in the center of the eye surrounding the pupil), may also be the first signs of retinoblastoma.
Diagnosis
The diagnosis of retinoblastoma is frequently made by the parents of an infant or child with the condition. Often, the parents will tell the physician that they have noticed that their child’s eye looks “white,” or that the child’s eye or eyes seem to drift to one side or the other.
When a child is born into a family that has a history of retinoblastoma, diagnosis of the condition can often be made before the baby leaves the hospital by an eye specialist, or ophthalmologist. If there is no family history, and the initial diagnosis is made by the parents or family physician, then the child can be sent to an ophthalmologist for a more thorough eye exam.
To examine a child for retinoblastoma, dilating drops are placed in both eyes to dilate (enlarge) the pupils and allow the ophthalmologist to view the retina. If a tumor is seen or suspected, an ultrasound examina-
tion, which uses sound waves to penetrate and outline structures in the eye, is used to confirm the presence of a tumor. A specialized x ray, called a CAT scan, which uses computers to take very detailed pictures of the inside of the body, can be used to see if there are tumors in other parts of the body.
Treatment and management
Treatment options for retinoblastoma have significantly increased over the past twenty years. The earliest form of treatment for retinoblastoma was enucleation, the removal of the major portion of the affected eye. This led to total loss of vision in that eye. While enucleation is still used, especially when the tumor is very large, newer, more sophisticated treatments have emerged that offer the chance to save at least some vision in the affected eye.
Lasers can be used in a treatment known as photocoagulation. This treatment is best used when the tumor is small and confined to the retina. The laser is actually used to burn and destroy blood vessels that feed the tumor, rather then directly on the tumor itself. The treatment can be repeated one or two times, and in some studies complete remission of the retinoblastoma was achieved in 70% of patients.
Another modality that works well with tumors that are confined to the retina is cryotherapy. It may be used as either the primary mode of treatment or in conjunction with other treatment modalities. Like photocogulation, cryotherapy has its highest success rates with smaller tumors. Unlike photocoagulation, cryotherapy uses extreme cold to destroy the tumor itself.
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
993 |
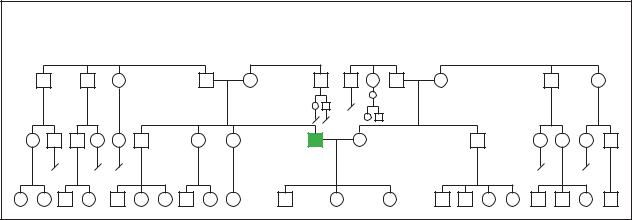
Retinoblastoma |
Retinoblastoma |
Sporadic |
|
|
Pedigree analysis showing sporatic occurance of retinoblatoma within a family. (Gale Group)
Thermotherapy uses heat generated from ultrasound or microwaves to destroy the retinoblastoma tumor. While thermotherapy works well on its own with small tumors, it is even more effective when used with chemotherapy or radiation therapy, which are thought to make the tumor more susceptible to the heat generated by thermotherapy.
The use of conventional external beam radiation is still used for retinoblastoma, especially for tumors that are larger and have spread outside the retina. While radiation is applied directly to the tumor, with careful application the eye itself can be saved from destruction in about 75% of patients. In 35% of patients who receive external beam radiation, there is an increased risk for a retinoblastoma tumor to develop in the other eye within a 30-year time frame. Therefore, external beam radiation is generally only used when other conservative measures, such as cryotherapy or photocoagulation fail or cannot be used due to large tumor size.
Probably the most significant advancement in the treatment and management of retinoblastoma has come about in the use of chemotherapy. While in the past chemotherapy was only used to treat patients whose tumors had spread outside the eye, newer chemotherapy agents such as carboplatin and etoposide, along with older agents such as vincristine, are being used to treat tumors that are confined to the eye with significant success. Using these chemotherapeautic agents, it has been shown that tumors typically decrease in size 30–45%. This then allows more conservative and eye-sparing therapy such as cryotherapy and photocoagulation to be used much more effectively.
Prognosis
The prognosis for the vast majority of patients with retinoblastoma is excellent. In the United States, over
95% of children with retinoblastoma survive and lead healthy, productive lives.
Children who have unilateral retinoblastoma have at least one normal eye and can lead normal childhood lives, and even drive cars as they get older. The majority of children with bilateral retinoblastoma retain some vision in one eye, and sometimes both eyes. However, all children affected with bilateral retinoblastoma and 15% of children with familial unilateral retinoblastoma have a higher risk of developing other cancers throughout their lives. Therefore, children in these categories need to have regular medical checkups throughout their lives to watch for any signs of secondary cancers in areas such as bone, muscle, skin, and brain.
Resources
BOOKS
Gelehrter, T., F. Collins, and D. Ginsburg. Principles of Medical Genetics. Baltimore, MD: Williams and Wilkins, 1998.
PERIODICALS
Benz, Matthew, Ingrid Scott, Timothy Murray, Deborah Kramer, and Stuart Toledano. “Complications of Systemic Chemotherapy as Treatment of Retinoblastoma.” Archives of Ophthalmology 118 (April 2000): 577-578.
MacDonald, Deborah, and Mira Lessick. “Hereditary Cancers in Children and Ethical and Psychosocial Implications.”
Journal of Pediatric Nursing 15, no. 4 (August 2000): 217220.
Zhao, Da-You, Carol Shields, Jerry Shields, and Kann Gunduz. “New Developments in the Management of Retinoblastoma.” Journal of Ophthalmic Nursing and
Technology 17, no. 1 (1998): 13-18.
ORGANIZATIONS
Eye Cancer Network. 115 East 61st St., New York, NY 10021. (212) 832-8170. http://www.eyecancer.com .
994 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
National Eye Institute. Bldg. 31 Rm 6A32, 31 Center Dr., MSC 2510, Bethesda, MD 20892-2510. (301) 496-5248. 2020@nei.nih.gov. http://www.nei.nih.gov .
National Retinoblastoma Research and Support Foundation. PO Box 016880, 900 NW 17th St., Room 257, Miami, FL 33101-6880. (800) 226-2734.
University of Pennsylvania Cancer Center. 3400 Spruce St., Philadelphia, PN 19104. (215) 662-4000. www.oncolink
.upenn.edu .
Edward R. Rosick, DO, MPH, MS
Retinoic acid embryopathy see Accutane embryopathy
I Rett syndrome
Definition
Rett syndrome is a progressive neurological disorder seen almost exclusively in females. The most common symptoms include decreased speech, mental retardation, severe lack of coordination, small head size, and unusual hand movements.
Description
Dr. Andreas Rett first reported females with the symptoms of Rett syndrome in 1966. Females with this X-linked dominant genetic condition are healthy and of average size at birth. During infancy, head growth is abnormally slow and microcephaly (small head size) develops. Babies with Rett syndrome initially have normal development. At approximately one year of age, development slows and eventually stops. Patients with Rett syndrome develop autistic features. Involuntary hand movements are a classic feature of Rett syndrome.
Females with Rett syndrome may also develop seizures, curvature of the spine (scoliosis), irregular breathing patterns, swallowing problems, constipation, and difficulties walking. Some females with Rett syndrome are unable to walk. There is currently no cure for Rett syndrome. Most girls with Rett syndrome live until adulthood. The gene responsible for Rett syndrome has been identified and genetic testing is available.
Genetic profile
Rett syndrome is an X-linked condition. This means that the mutation (genetic change) responsible for Rett syndrome affects a gene located on the X chromosome.
K E Y T E R M S
Apraxia—Impairment of the ability to make purposeful movements, but not paralysis or loss of sensation.
Ataxia—A deficiency of muscular coordination, especially when voluntary movements are attempted, such as grasping or walking.
Autism—A syndrome characterized by a lack of responsiveness to other people or outside stimulus. Often in conjunction with a severe impairment of verbal and non-verbal communication skills.
Microcephaly—An abnormally small head.
Mutation—A permanent change in the genetic material that may alter a trait or characteristic of an individual, or manifest as disease, and can be transmitted to offspring.
Neuron—The fundamental nerve cell that conducts impulses across the cell membrane.
Scoliosis—An abnormal, side-to-side curvature of the spine.
Spasticity—Increased muscle tone, or stiffness, which leads to uncontrolled, awkward movements.
The affected gene is the methyl CpG-binding protein 2 (MECP2) gene. This gene makes a protein that regulates other genes. When there is a mutation in MECP2, the protein it makes does not work properly. This is thought to prevent normal neuron (nerve cell) development.
Rett syndrome is considered to be X-linked dominant in nature. Males have one X chromosome and one Y chromosome. Females have two X chromosomes. Males with a mutation in their MECP2 gene typically die as infants or are miscarried before birth. Rett syndrome is usually considered fatal in males because the Y chromosome cannot compensate for the MECP2 mutation on the X chromosome. Females with a mutation in the MECP2 gene develop Rett syndrome, but the presence of the second X chromosome in females carrying a normal MECP2 gene enables them to survive.
The severity of the syndrome in females is related to the type of mutation in the MECP2 gene and the activity of the X chromosomes. Normally, both X chromosomes have the same activity. However, the activity can be unequal. If the X chromosome with the mutation in the MECP2 gene is more active than the X chromosome without the mutation, the female is more severely
syndrome Rett
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
995 |
Rett syndrome
affected. The reverse is also true. If the X chromosome without the mutation is more active than the X chromosome with the mutation, the female is less severely affected.
If a woman has a mutation in her MECP2 gene, she has a 50% risk with any pregnancy to pass on her X chromosome with the mutation. However, it is uncommon for women with Rett syndrome to have children due to the severity of the disorder.
Demographics
The incidence of Rett syndrome is thought to be between 1 in 10,000 and 1 in 20,000 live births. It is seen almost exclusively in females. The vast majority of cases of Rett syndrome are sporadic in nature. Therefore, the risk of a family having more than one affected daughter is typically very low.
Signs and symptoms
Infants with Rett syndrome typically have normal size at birth. They develop normally until approximately 6–18 months of age. Development then slows, eventually stops, and soon regresses. Affected individuals are unable to do things they were once able to do. Girls with Rett syndrome lose the ability to speak, become uninterested in interacting with others, and stop voluntarily using their hands. The loss of language and eye contact causes girls with Rett syndrome to appear to be autistic. Between one and three years of age, girls with Rett syndrome develop the unusual hand movements that are associated with the disease. Patients wring their hands, clap their hands, and put their hands in their mouth involuntarily. Some patients with Rett syndrome also lose the ability to walk. If the ability to walk is maintained, the gait is very ataxic (uncoordinated, clumsy).
By preschool age the developmental deterioration of girls with Rett syndrome stops, but they continue to have lack of speech, inability to understand language, poor eye contact, mental retardation, ataxia, and apraxia (inability to make purposeful movements). Other common symptoms associated with Rett syndrome include seizures, constipation, irregular breathing, scoliosis, swallowing problems, teeth grinding, sleep disturbances, and poor circulation. As patients with Rett syndrome get older, their ability to move decreases and spasticity (rigidity of muscles) increases.
Diagnosis
The diagnosis of Rett syndrome is made when the majority of the symptoms associated with the disease are
present. If a physician suspects an individual has Rett syndrome, DNA testing is recommended. Approximately 75% of patients with Rett syndrome have a mutation in the MECP2 gene. DNA testing can be performed on a blood sample, or other types of tissue from the body. If a mutation is found in the MECP2 gene, the diagnosis of Rett syndrome is confirmed.
Treatment and management
As of 2001, there is not a cure for Rett syndrome. Treatment of patients with Rett syndrome focuses on the symptoms present. Treatment may include medications that inhibit seizures, reduce spasticity, and prevent sleep disturbances. Nutrition is monitored in females with Rett syndrome due to their small stature and the constipation associated with the disorder.
Prognosis
In the absence of severe medical problems, most patients with Rett syndrome live into adulthood.
Resources
BOOKS
Zoghbi, Huda, and Uta Francke. “Rett Syndrome.” In The
Metabolic and Molecular Bases of Inherited Disease. New
York: McGraw-Hill, 2001.
PERIODICALS
Percy, Alan. “Genetics of Rett Syndrome: Properties of the Newly Discovered Gene and Pathophysiology of the Disorder.” Current Opinion in Pediatrics 12 (2000): 589595.
ORGANIZATIONS
Alliance of Genetic Support Groups. 4301 Connecticut Ave. NW, Suite 404, Washington, DC 20008. (202) 966-5557. Fax: (202) 966-8553. http://www.geneticalliance.org .
International Rett Syndrome Association. 9121 Piscataway Rd., Clinton, MD 20735. (800) 818-RETT. http://www
.rettsyndrome.org .
National Organization for Rare Disorders (NORD). PO Box 8923, New Fairfield, CT 06812-8923. (203) 746-6518 or (800) 999-6673. Fax: (203) 746-6481. http://www
.rarediseases.org .
Rett Syndrome Research Foundation. 4600 Devitt Dr., Cincinnati, OH 45246. http://www.rsfr.org .
WEBSITES
“Rett Syndrome.” Online Mendelian Inheritance in Man.
http://www. ncbi . nlm . nih . gov/entrez/dispomim
.cgi?id=312750 . (May 24, 2000).
Holly A. Ishmael, MS, CGC
RFH1 see Renal failure due to hypertension
996 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |

I Rhizomelic chondrodysplasia punctata
Definition
Rhizomelic chondrodysplasia punctata is a rare, severe, inherited disease. The main features are limb shortening, bone and cartilage abnormalities visible on x ray, abnormal facial appearance, severe mental retardation, profound psychomotor retardation, and cataracts. Skeletal abnormalities can be seen prenatally. Most affected persons die in infancy. No treatments are available.
Description
Rhizomelic chondrodysplasia punctata (RCDP) is caused by an abnormal protein in a part of the cell called the peroxisome. The inside of the cell contains compartments (called “organelles”) that perform specific functions. The peroxisome functions in many metabolic processes, especially those involving lipids (fats) and hydrogen peroxide. Multiple peroxisomes are in almost every human cell. RCDP is one of many peroxisomal disorders, as well as a metabolic disorder.
Three other conditions are also called “chondrodysplasia punctata.” These conditions are different from RCDP. They have almost the same name because it describes a feature that is present in all four conditions. However, the causes, features, and patterns of inheritance of the other chondrodysplasia punctata conditions are different from those of RCDP.
Genetic profile
Rhizomelic chondrodysplasia punctata is an autosomal recessive condition. This means that it occurs in both males and females, and often affects people who have no family history of the condition. Humans have two copies of every gene, one maternally and one paternally inherited. Autosomal recessive conditions occur when a person has two abnormal copies of the same gene. People who have one abnormal copy and one normal copy of a particular gene are unaffected; they are called “carriers.” An affected person has inherited two abnormal RCDP genes, one from each carrier parent. The risk for the carrier parents to have another affected child is then 25% with each pregnancy.
In 1997, the gene that causes RCDP was identified. The gene is called PEX7 and it is on chromosome 6. Fifteen genes involved in the synthesis of peroxisomes have been identified in humans. These genes are called PEX genes, and the proteins they code for are called peroxins. Disorders caused by abnormalities of peroxin proteins are often called “peroxisomal biogenesis” disorders.
The PEX7 gene codes for a peroxisomal component that helps transport other important proteins into the peroxisome. The proteins to be transported contain a signal, called “PTS2” (peroxisome targeting sequence 2) that is recognized by the receptor on the peroxisome. When PEX7 is abnormal, the receptor that usually recognizes and helps transport the PTS2 proteins is abnormal. Thus, the abnormality of this one receptor has a cascade effect on many other proteins.
Demographics
Rhizomelic chondrodysplasia punctata is quite rare. It occurs in fewer than 1/100,000 births. The incidence of peroxisomal biogenesis disorders is approximately 1/50,000 births; RCDP accounts for fewer than one fifth of these.
Signs and symptoms
“Rhizomelic” refers to shortening of the bones near the center of the body (the bones of the thighs and upper arms more so than the bones of the forearms and lower legs). “Chondro” refers to cartilage and “dysplasia” to abnormal development. “Punctata” refers to specific abnormalities seen on radiological studies such as x ray. The ends of the bones near joints appear to be spotted. The spots represent dense, abnormal cartilage. The spots are also called “punctate calcifications.” Other abnormalities include frozen joints (called contractures), abnormal facial features, cataracts, hearing loss, severe mental retardation, and profound psychomotor retardation. People with RCDP may also have other bone abnormalities, small heads, coarse and sparse hair, and dry, red skin.
The proximal shortening of the bones causes short stature, which is apparent before birth. Growth after birth is retarded as well. The rhizomelic shortening is severe, and occurs to the same degree on both sides of the body. The stippling (spotting) of the bones mainly involves the ends of the bones near the hip, knee, elbow, and shoulder. “Severe” mental retardation describes cognitive deficits worse than those of typical Down syndrome. Some researchers have described degeneration of brain tissue after birth. Researchers are not sure of the reason for this; it may be due to toxic effects of excess phytanic acid. Cataracts are symmetrical and occur in both eyes. The abnormal facial features have been called “koala bear facies.” Facial features include a broad forehead and a saddle nose.
A subset of people with RCDP do not have some of the typical symptoms, such as shortening of proximal bones and/or severe mental retardation. The diagnosis in these individuals was confirmed to be RCDP. Therefore, the spectrum of features in RCDP is variable;
punctata chondrodysplasia Rhizomelic
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
997 |

Rhizomelic chondrodysplasia punctata
K E Y T E R M S
Anticoagulant—Drugs used to prevent blood clots.
Cell—The smallest living units of the body which group together to form tissues and help the body perform specific functions.
Differentiate—Specialized development to perform a particular function.
Gene—A building block of inheritance, which contains the instructions for the production of a particular protein, and is made up of a molecular sequence found on a section of DNA. Each gene is found on a precise location on a chromosome.
Metabolism—The total combination of all of the chemical processes that occur within cells and tissues of a living body.
Plasmalogens—Fat molecules that are important components of cells and of the myelin sheath that protects nerve cells.
Protein—Important building blocks of the body, composed of amino acids, involved in the formation of body structures and controlling the basic functions of the human body.
Psychomotor—Movement produced by action of the mind or will.
some people are much more mildly affected than is typical. These differences in severity appear to be associated with different mutations in the PEX7 gene.
Diagnosis
Although suspicion of RCDP is raised by the physical and radiographic features, the diagnosis is made by laboratory testing. People with RCDP have very specific biochemical abnormalities, i.e. abnormal levels of particular substances in bodily fluids. These abnormalities are due to the underlying defect in the peroxisome. The specific abnormalities are: 1) deficient plasmalogen synthesis with very low plasmalogen levels in red blood cells, 2) inability to process (oxidize) phytanic acid leading to elevated levels of phytanic acid in the blood, and 3) an unprocessed form of peroxisomal thiolase. Phytanic acid levels are normal at birth and increase to at least ten times normal by one year of age. Some experts recommend that confimatory studies be performed on cells obtained by skin biopsy.
The biochemical studies diagnostic of RCDP can be performed prenatally on cells obtained by chorionic vil-
lus sampling (CVS) or amniocentesis. CVS is usually performed at 10–12 weeks of pregnancy and amniocentesis is usually performed after 15 weeks of pregnancy. RCPD may be suspected in a fetus based on ultrasound findings.
Each feature of RCDP is seen in many other conditions, for example rhizomelic limb shortening is seen in other conditions that cause dwarfism. Chondrodysplasia punctata is seen in many inherited conditions but can also be caused by prenatal exposure to the anticoagulant drug, Warfarin. Doctors who specialize in diagnosing rare genetic conditions use subtle differences between the symptoms of these conditions to narrow their search for the suspected diagnosis. Many peroxisomal disorders have abnormal very long chain fatty acids (VLCFAs); VLCFA levels are normal in RCDP.
Two rare conditions cannot be distinguished from RCDP by physical symptoms. These conditions involve specific abnormalities of plasmalogen synthesis. RCDP is caused by abnormal peroxisome synthesis, which leads to multiple biochemical abnormalities including deficient plasmalogen synthesis. In contrast, these two conditions each affect only one protein. The proteins affected are dihydroxyacetone phosphate acyltransferase (DHAPAT) and alkyl dihydroxyacetone phosphate synthase. People with deficiencies in these two proteins have normal thiolase and normal phytanic acid levels.
RCDP is the only condition known to be caused by abnormal PEX7 gene. Genetic testing is another method to confirm the diagnosis. A doctor who specializes in medical genetics can determine whether this testing is available clinically.
Treatment and management
The only treatments for RCDP are supportive therapies to treat symptoms. People with RCDP, especially those who are less severely affected, benefit from symptomatic support of various specialties such as ophthalmology and physical therapy. Dietary restrictions or supplements have shown promise in the treatment of some peroxisomal disorders. The enormous obstacle in the severe conditions is that many of the abnormalities develop before birth and are irreversible. The multiple biochemical abnormalities of RCDP also complicate treatment efforts. Some researchers have tried to improve the function of the deficient metabolic process. This treatment, if it works, will probably benefit mildly affected patients more than the typically severely affected person with RCDP.
The underlying cause of the severe mental retardation is not well understood. Some abnormalities of nerve tissue have been described. In many peroxisomal disor-
998 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |

ders similar to RCDP the nerve tissue migrates abnormally before birth. This abnormal migration is not present in RCDP. It appears that in RCDP the nerve tissue does not differentiate properly once it has migrated to the correct location in the body.
Prognosis
The prognosis for the typical individual with RCDP, who is severely affected, is death in infancy. Most affected infants die in the first two years of life. However, exceptions have reported in the medical literature. Individuals who lived past the age of 10 years have been reported. For atypical, mildly affected patients, prognosis is variable.
Scientists’ understanding of peroxisomal disorders, and of the peroxisome itself, increased enormously in the last five years. Developing effective treatments of RCDP is a great challenge. But having a better understanding of the underlying cause is the first step. This has also increased awareness of RCDP, probably leading to more accurate diagnoses and higher clinical suspicion. A correct diagnosis is critical in providing accurate recurrence, prognosis, and prenatal diagnosis information.
Resources
PERIODICALS
Bennett, Ruth. “Workshop Looks Into the Challenges, Causes of Dwarfism.” The Oregonian (July 7, 1999).
Hedley, Lisa Abelow. “A Child of Difference.” New York Times Magazine 147 (October 1997): 98-99.
ORGANIZATIONS
Footsteps Institute for Rare Diseases. 624 Martin Luther King Way, Tacoma, WA 98405. (253) 383-0985 or (888) 6404673. rwrfsi@aol.com.
International Patient Advocacy Association. 800 Bellevue Way NE, Suite 400, Bellvue, WA 98004. (425) 462-4037 or (310) 229-5750 or (800) 944-7823 x4037. lvp.ipaa@att
.net. http://www.vanpelt-ipaa.com .
National Organization for Rare Disorders (NORD). PO Box 8923, New Fairfield, CT 06812-8923. (203) 746-6518 or (800) 999-6673. Fax: (203) 746-6481. http://www
.rarediseases.org .
Rhizomelic Chondrodysplasia Punctata (RCP) Family Support Group. 137 25th Ave., Monroe, WI 53566.
WEBSITES
“Genetics Tutorial and diagnosis information.” Greenberg Center for Skeletal Dysplasias. Johns Hopkins University.http://www.med.jhu.edu/Greenberg.Center/Greenbrg
.htm .
Gould, Stephen J. “The Peroxisome Website.” John Hopkins University School of Medicine. http://www.peroxisome
.org .
Michelle Queneau Bosworth, MS, CGC
I Rhodopsin
Definition
Rhodopsin is the visual pigment that “senses” light in the rod cells of the retina.
Where is rhodopsin?
Rhodopsin is found at the back of the eye, in the retina. The retina is the area of the eye that senses light, interprets that information, and transmits it to the brain for further interpretation. Two types of light-sensing cells are found in the retina: rods and cones. In a simplified explanation, rod cells are responsible for black and white vision, whereas cone cells are responsible for color vision. This is true as far as it goes, but there are many more differences between rods and cones.
In rod cells, rhodopsin is responsible for phototransduction, the process of turning light into chemical and electrical energy. Rhodopsin is responsible for phototransduction in rod cells, but not in cone cells. Three different proteins, similar to rhodopsin, govern phototransduction in the cone cells. Each of these three phototransducers responds to a different color of light, which allows persons with normal color vision to see the entire color spectrum.
In order to understand more of the structure, function, and location of rhodopsin, a discussion of cells and cell membranes is necessary. Every human cell has a cell membrane that separates the environment inside the cell (intracellular environment) from the extracellular (outside the cell) environment. Cell membranes are made up of lipids, which are hydrophobic substances. Hydrophobic literally means “fear of water.” Oil is an example of a hydrophobic substance. If oil is added to water, the oil will separate itself from the water. Basically, the lipids in the cell membrane form a similar water-excluding ball, but the inside of the ball will contain water (and other intracellular fluids). Each rhodopsin molecule crosses the cell membrane seven times, and each area of the protein in the cell membrane is called a transmembrane domain. These transmembrane domains (which are hydrophobic) dictate an interesting structure for rhodopsin. Imagine folding a hose seven times to hold it in your hand. The structure for rhodopsin is at least that complex. One reason to mention that rhodopsin has the seven transmembrane domains is because that structure is common to G proteins, and rhodopsin is a G protein. G proteins are generally involved in a biological cascade. A biological cascade is a system where a small initial input (like a brief flash of light) can result in a rather large output.
Rhodopsin
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
999 |
Rhodopsin
How does rhodopsin turn light into a chemical signal?
Rhodopsin is a combination of two different molecules, retinal and opsin. Retinal is a derivative of vitamin A, and opsin is a protein. When rhodopsin is not activated, retinal is in the 11-cis configuration. When light hits 11-cis retinal, it changes its shape to become all-trans retinal. This is the only light-sensitive step in vision (in the rod cells). What the configurations are called, and what those names mean is not as important as the fact that this light-dependent change in conformation results in light being converted into chemical energy.
Once retinal reaches the all-trans conformation, opsin also changes its shape. The new opsin-retinal complex is called metarhodopsin II. Metarhodopsin II is a semistable complex that is the active form of rhodopsin. Metarhodopsin II, unlike the inactive rhodopsin, is able to bind a protein called transducin. Each metarhodopsin II can bind to many transducins (literally hundreds). These transducins then cause a decrease in cGMP concentration, and one transducin molecule can cause the breakdown of more than 1,000 cGMP per second. One can clearly see why the G protein cascades are excellent systems for amplifying a signal.
Mutations in rhodopsin
Mutations in rhodopsin can result in two different disorders—retinitis pigmentosa and congenital stationary night blindness. Retinitis pigmentosa (RP) affects about one in 3,000 persons living in the United States, and about 1.5 million persons worldwide. Many mutations, not just mutations of the rhodopsin gene, lead to RP. The disorder may be inherited in an X-linked recessive fashion in 8% of all cases, an autosomal dominant fashion in 19% of cases, or as an autosomal recessive disorder in 19% of all cases. In the rest of the cases (54%), the mutations do not follow classical genetic patterns of inheritance. Mutations in rhodopsin have been found to cause approximately 20% of the autosomal dominant form of RP. The rhodopsin gene is located at the 3q locus of chromosome 3.
Patients with retinitis pigmentosa exhibit symptoms that include night blindness, abnormal pigment accumulation in the retina, and a progressive decrease in the visual fields. The patient’s vision decreases from the outermost edges in. The age of onset of the disorder may be as young as six months, but most patients experience the first symptoms between ages 10 and 30. In RP, the patient’s rod cells usually degenerate first, followed by a loss of cone cells.
Symptoms may often present after a motor vehicle accident. Not only is the age of onset variable, but the severity of the disease is as well. Patients with the same mutation, even within the same family, exhibit differing severities of the disorder. Mutations in rhodopsin may also cause autosomal recessive cases of RP.
Congenital stationary night blindness (CSNB) is another disorder that can be caused by mutations in the rhodopsin gene. Patients with CSNB, as may be deduced from the name, experience night blindness. However, unlike RP, patients with CSNB do not experience degeneration (death) of cells of the retina (rod and cone cells). Patients with CSNB are thought to have an overactive transducin molecule, which prevents their rods from functioning normally. A mutation in transducin, which also causes CSNB, supports this theory, since this transducin is also thought to be overly active.
Treatment
As of 2001, no effective treamtment for RP exists. However, new treatments are being explored for RP. Experiments in rats have shown that rod cells can be affected by gene therapy. Although gene therapy has not been successfully demonstrated as of this printing, at least the hope now exists that eventually gene therapy may be applied to the problem of RP. Previously, addition of a new gene into a non-dividing cell line had been thought to be technically insurmountable. Another experiment in rodents offers hope for those who have autosomal recessive RP. In rats with autosomal recessive RP, retinal pigment transplantation has successfully treated them according to Columbia University’s Retinal Transplant newsletter. This technique might prove promising in humans.
Prognosis
The prognosis for persons with RP is extremely variable. Persons with CSNB will experience night blindness throughout their lives.
Resources
WEBSITES
“A guide to retinitis pigmentosa.” The British Retinitis Pigmentosa Society. http://www.brps.demon.co.uk/ Graphics/G_Guide.html#SYMPTOMS .
Robbins, Alexandra. “Congenital stationary night blindness.”http://130.132.19.190/thom/reviews/cnsb.html .
Michael V. Zuck, PhD
1000 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |

Rhodopsin related retinitis pigmentosa see
Rhodopsin
Rhymes syndrome see Retinitis pigmentosa
Ribonucleic acid see RNA
RIEG see Rieger syndrome
I Rieger syndrome
Definition
Rieger syndrome is a rare disorder characterized by absence and/or malformation of certain teeth, mild craniofacial (relating to the head and the face) abnormalities, and various eye abnormalities. The eye abnormalities, referred to as Rieger eye malformations, may be present separately or as a part of Rieger syndrome.
Description
First characterized by Herwigh Rieger, an Austrian ophthalmologist in 1935, Rieger syndrome is a dominantly inherited disease. Disease expression is highly variable, including craniofacial, ocular, and dental malformations. Symptoms may also include myotonic dystrophy (a condition characterized by delay in the ability to relax muscles), umbilical abnormalities (abnormalities relating to where the umbilical cord attaches to a baby), and other defects. Psychomotor retardation, a slowing of the motor action directly proceeding from mental activity, occurs in some cases.
Rieger syndrome is also sometimes referred to as goniodysgenesis hypodontia, iridogoniodysgenesis with somatic anomalies, or RGS. It is a multiple congenital anomaly syndrome, a syndrome marked by multiple abnormalities at birth. Currently, there are two genetic types of Rieger syndrome identified. Type I results from a mutation on chromosome 4 and Type II on chromosome 13.
Genetic profile
Rieger syndrome is inherited as an autosomal dominant disease. In autosomal dominant inheritance, a single abnormal gene on one of the autosomal chromosomes (one of the first 22 non-sex chromosomes) from either parent can cause the disease. One of the parents will have the disease (since it is dominant) and is the carrier. Only one parent needs to be a carrier in order for the child
to inherit the disease. A child who has one parent with the disease has a 50% chance of also having the disease.
There is evidence that there is more than one genetic form of Rieger syndrome. The disease gene responsible for Rieger syndrome Type I is caused by mutations in the RIEG1 gene, which is located on the long arm (q) of chromosome 4 (4q25-Q26).
Linkage studies have indicated that a second type of Rieger syndrome, Type II, maps to the long arm of chromosome 13, at 13q14 (gene RIEG2).
Demographics
Rieger syndrome is very rare. Little is known in regard to the number of affected individuals or whether certain areas or ethnic groups are at a greater risk. Since the disease appears to be inherited in an autosomal dominant manner, meaning that it is transmitted on one of the non-sex chromosomes, males and females have an equal chance of acquiring the abnormal gene from their parents.
Signs and symptoms
The symptoms of Rieger syndrome are expressed variably. The main symptoms of Rieger syndrome are:
•Ocular malformations, called Rieger eye malformations, include underdeveloped iris, a small cornea (microcornea), an opaque ring around the outer edge of the cornea, adhesions (abnormal union of surfaces normally separate) in the front of the eye, and/or displacement of the pupil of the eye so that it is not centered.
•Dental abnormalities include a congenital condition causing a fewer number of teeth than normal (hypodontia); a condition in which a single tooth, pairs of teeth, or all the teeth are smaller than normal (microdontia), and/or cone-shaped.
•Craniofacial abnormalities include a protruding lower lip, a broad, flat bridge of the nose, and/or underdeveloped bones of the upper jaw (hypoplasia) causing the face to have a flat appearance.
Other conditions that have been found in some patients with Rieger syndrome are:
•Anal stenosis (a small anal opening).
•Failure of the skin around the navel to decrease in size after birth.
•Protrusion of intestine through a weakness in the abdominal wall around the navel (umbilical hernia).
•Glaucoma (increased pressure within the eyeball) may result from the ocular malformations associated with Rieger syndrome, including defects in the angle of the eye that is created by the iris and cornea (trabeculum),
syndrome Rieger
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
1001 |