

ТОХТ
.pdfЕсли скорость обрыва цепи больше скорости продолжения цепи, то цепной процесс не развивается и реакция протекает по радикальному нецепному механизму:
|
|
|
Cl |
2 |
k0 |
2 Cl . |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
k |
|
. |
|
|
C H CH |
3 |
Cl. |
1 |
C H CH |
2 |
HCl |
|||
6 |
5 |
|
|
|
6 |
5 |
|
||
|
. |
|
|
k |
|
|
|
C H CH |
|
Cl . |
2 |
C H CH Cl |
|||
2 |
|
||||||
6 |
5 |
|
|
6 |
5 |
2 |
|
В этом случае скорость нецепного хлорирования толуола равна
[ ],
т.е. скорость лимитируется стадией образования атомов хлора. 13.8. Кинетика разветвленных цепных реакций
Разветвленные цепные реакции (к ним относится окисление) отличаются от неразветвленных тем, что они имеют дополнительные источники образования свободных радикалов: 1) превращения свободных радикалов, участвующих в продолжение цепи, с образованием большего числа радикалов и 2) реакции молекулярных продуктов, ведущие к образованию новых радикалов (так называемое вырожденное разветвление цепи).
При газофазных реакциях окисления вырожденное разветвление цепи обычно происходит на образующихся альдегидах, которые достаточно легко реагируют с кислородом
Примером цепной вырождено-разветвленной реакции является окисление углеводородов в гидроперекиси молекулярным кислородом в жидкой фазе, протекающее по схеме:
- зарождение цепи
|
|
|
|
k |
|
|
. |
|
|
|
|
|
. |
|
RH |
|
O |
0 |
|
|
|
HOO |
|||||||
|
|
|
|
R |
|
|||||||||
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
- продолжение цепи |
|
|
|
|
|
|
|
|
||||||
|
|
|
k |
|
|
|
|
|
|
|
|
|
|
|
R. |
|
O |
1 |
|
ROO. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
k |
|
|
|
|
|
R . |
|
|||
ROO . |
RH |
|
2 |
ROOH |
|
|||||||||
|
|
|
||||||||||||
- вырожденное разветвление цепи |
|
|||||||||||||
ROOH |
k3 |
|
RO. |
|
HO. |
|
||||||||
|
k |
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
RO . |
|
|
|
|
2 ROOH |
4 |
|
ROO . |
|
|
|
|
H O |
||||||
|
|
|
||||||||||||
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2 |
Скорость зарождения цепи по реакции (1) как правило мала по сравнению со скоростью вырожденного разветвления цепи. По мере накопления в окисляемом углеводороде пероксида скорость инициирования будет определяться скоростью распада гидропероксида на радикалы. По этой причине наблюдается довольно длительный индукционный период, который можно сократить, добавив инициаторы, в том числе гидроперекись, образующуюся в этом же процессе, или «затравку» уже окисленной реакционной массы от предыдущей операции.
221

Несмотря на малую реакционную способность перекисных радикалов, небольшая растворимость кислорода в жидкости делает возможным разные способы квадратичного обрыва цепи:
При обрыве цепи на перекисных радикалах получим выражение для скорости окисления:
√ [ ]
√
а при обрыве на углеводородных радикалах:
√ [ ]
√
Последнее уравнение, соответствующее первому порядку по кислороду, соблюдается при его низких парциальных давлениях. При повышении давления более 133 кПа (более 100 мм рт. ст.) порядок по кислороду постепенно снижается и становится нулевым (рис. 13.4), что указывает на изменение способа обрыва цепи.
Рис 13.4. Зависимость скорости окисления от парциального давления кислорода
Скорость зарождения цепи, входящая в предыдущие уравнения, при термическом окислении складывается из трех слагаемых:
[ |
][ |
] |
[ |
] |
[ |
] |
Первое из них существенно только в начальной стадии реакции, а при стационарном процессе окисления оно столь мало, что им вполне можно пренебречь. Тогда при обрыве на перекисных радикалах получаем:
|
|
|
|
|
|
|
√ [ |
] |
[ |
] |
[ ] |
||
|
|
|
|
|
|
|
|
|
√ |
|
|
|
|
Это уравнение показывает автоускорение процесса с S-образной кривой накопления продуктов, характерной для разветвленных реакций (рис. 13.5).
222

Рис. 13.5. Зависимость концентрации продукта окисления от времени реакции
При целевом получении гидроперекисей их концентрацию в реакционной массе доводят до 10-30 вес. %. В других реакциях окисления гидроперекиси в ходе реакции разлагаются с образованием спиртов, кетонов или альдегидов.
Длина кинетической цепи равна отношению скоростей продолжения и обрыва цепи или инициирования:
√ |
|
[ ] |
|
|
[ ] |
|
|
|
|||
|
|
|
√ |
||
С увеличением скорости инициирования длина цепи уменьшается, что характерно для квадратичного обрыва цепи. Чем выше скорость образования свободных радикалов, тем выше их концентрация, тем чаще происходит рекомбинация двух свободных радикалов.
Процессы окисления чаще проводят в присутствии катализаторов.
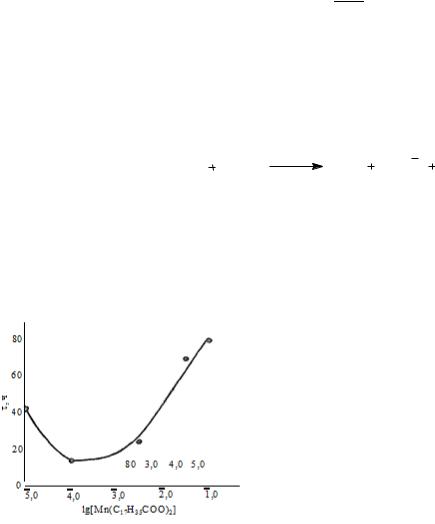
При окислении углеводородов в присутствии металлов переменной валентности повышается скорость процесса, изменяется состав продуктов окисления, иногда существенно меняется режим окисления или цепного процесса.
Каталитическое действие солей металлов переменной валентности (Mn2+, Co2+, Fe2+) обусловлено ускорением реакций вырожденного разветвления цепи при разложении перекисных соединений. Более точно установлена схема превращений для окисления альдегидов:
1) RCHO |
|
3+ |
k |
0 |
RCO . |
2+ |
+ |
|
|
||||||
|
Co |
|
|
Co |
H |
||
|
|
k |
|
|
|
|
|
2) RCO . |
O |
1 |
|
|
RCO |
|
|
2 |
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
OO. |
|
|
3) RCO |
|
RCHO |
k2 |
|
RCOOOH |
|
RCO . |
|
|
|
|
|
|
|
|||||
|
|
|
|
||||||
OO. |
|
|
|
|
|
|
|
||
|
|
|
|
|
k |
RCOO. |
|
|
|
4) RCOOOOH |
Co2+ |
|
3 |
|
HO |
Co3+ |
|||
|
|
|
|||||||
Реакции (2) и (3) представляют собой элементарные стадии продолжения цепи. Обрыв цепи происходит квадратично на перацильных радикалах RCOOO· по реакции (5). Вырожденное разветвление цепи (4) при участии металла в его низшей валентной форме происходит значительно быстрее, чем тер-
223
 Co
Co ROO
ROO 
 Co
Co
3.Каким образом бромистый водород может катализировать реакции окисления? Объясните два возможных механизма этого явления, в том числе за счет участия НВrв стадиях продолжения цепи.
4.Объясните причины различий в среднем молекулярном весе теломеров при теломеризации четыреххлористого углерода с этиленом и стиролом.
5.Почему примесь пентана ингибирует реакцию термического дегидрохлорирования 1,2-дихлорэтана?
6.По какой причине 1,1,2,2-тетрахлорэтан отщепляет НС1 быстрее, чем 1,2- дихлортан, а последний — быстрее, чем 1,1-дихлорэтан?
7.При окислении углеводородов спирты получаются в тем большем количестве (по сравнению с кетонами), чем меньше парциальное давление кислорода. Объясните это явление.
8.Напишите элементарные реакции окисления изопропилбензола с образованием его гидроперекиси, диметилфенилкарбинола и ацетофенона. Какие из этих продуктов образуются параллельно, а какие — последовательно?
9.Как можно из этилбензола, пропилена и воздуха получить стирол и окись пропилена? Как получить окись пропилена и уксусную кислоту из пропилена, ацетальдегида и воздуха?
10.Каким образом из пропилена можно получить перв‒ и втор‒ пропилмеркаптан? Напишите реакции образования побочных продуктов ‒ тиоэфиров ‒ при радикально-цепной реакции.
11.Термический синтез фосгена в газовой фазе идет по механизму:
|
k0 |
2 Cl . |
|
. |
|
|
k |
|
|
Cl. |
|
1) Cl |
3) ClCO |
Cl |
|
COCl |
|
||||||
2 |
2 |
2 |
|||||||||
2 |
|
|
|
|
|
|
|
|
|
||
. |
|
k1 |
. |
|
. |
k |
|
|
|
|
|
CO |
ClCO |
4) |
|
Cl |
|
|
|
||||
2) Cl |
|
2 Cl |
t |
|
|
|
|
||||
|
|
|
|
|
|
|
|
2 |
|
|
|
Выведите кинетическое уравнение этой цепной реакции.
12. Какое кинетическое уравнение соблюдается при термическом крекинге пропана в случае линейного обрыва цепи на стенке реакционного сосуда?
13. Реакция термического гидродеалкилирования алкилбензолов протекает по следующему механизму:
CH |
|
|
. |
|
|
|
|
|
|
CH |
|
|
|
|
|
3 |
k |
|
|
|
|
|
|
|
0 |
2 |
. |
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
H |
|
|
|
CH3 |
|
k1 |
|
CH3 |
k2 |
. |
CH3 |
|
|
|
|||||
|
H. |
|
|
H |
|
C6H6 |
|
|
k-1 |
|
|
|
|
||
|
|
|
|
|
|
|
|
. CH3 H2 |
k3 |
CH |
H. |
|
2 H. kt |
H |
|
|
|
4 |
|
|
|
2 |
|
Каково кинетическое уравнение этого процесса?
225
Тема 14. Гомогенно-каталитические реакции
14.1. Двухстадийная реакция гомогенного катализа при одной лимитирующей стадии
По современным представлениям катализом называют изменение скорости химической реакции при воздействии вещества-катализатора, которое многократно участвует в элементарных химических реакций, но не входит в состав продуктов и не находится в стехиометрических отношениях с ними. Катализатор регенерируется после каждого цикла превращения реагентов в продукты и может расходоваться только по побочным реакциям.
В типичных случаях гомогенного катализа вначале по обратимой реакции образуется активный комплекс катализатора К с одним из реагентов (субстратом) А, причем эта стадия состоит в присоединении или замещении:
A |
K |
|
k |
1 |
AK |
A |
K |
k |
1 |
A' |
Z |
|
|
|
|||||||||
|
|
|
k |
|
|||||||
|
|
k |
-1 |
|
|
|
-1 |
|
|
||
|
|
|
|
|
|
|
|
|
|||
Затем образовавшийся комплекс (или активная частица) по бимолекулярной реакции взаимодействует со вторым реагентом или по мономолекулярной реакции распадается, с образованием молекулы продукта и регенерацией катализатора:
AK (A') |
Y |
k |
B |
K |
2 |
||||
AK (A') |
k |
B |
K |
|
2 |
||||
Возьмем для кинетического анализа наиболее общий случай, когда первая стадия состоит в замещении, а вторая является бимолекулярным взаимодействием. Скорость реакции описывается кинетическим уравнением:
[ ][ ]
где k2 ‒ истинная константа скорости каталитической реакции. Концентрацию активного комплекса (или частицы) находим из условия
стационарности
|
|
[ |
] |
|
[ |
][ |
] |
[ |
][ |
] |
[ ][ ] |
|||
откуда |
|
|
|
|
||||||||||
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
[ |
] |
[ |
][ |
] |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
[ |
] |
[ |
] |
|||
|
|
|
|
|
|
|
|
|
|
|||||
Подставляем значение [ |
] в уравнение скорости реакции и получаем: |
|||||||||||||
[ |
][ |
][ |
|
] |
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
||
[ |
] |
[ |
] |
|
|
|
|
|
|
|
||||
Если первая стадия сводится к присоединению, [Z] в уравнении исчезает, превращаясь в единицу. При мономолекулярности второй стадии то же происходит с [Y]. Аналогичные изменения наблюдаются, когда эти вещества находятся, в большом избытке или являются растворителями, что позволяет ввести, их концентрации в соответствующие константы скорости.
226

При анализе уравнения скорости представляют интерес два предельных случая:
1. Скорость превращения промежуточного комплекса на второй стадии значительно превышает скорость его обратного разложения на исходные вещества,, т., е. k2[Y] >> k-1[Z]. В этом случае величиной k-1[Z] в знаменателе можно пренебречь, а комплекс не может накопиться в заметном количестве. Если другие формы состояния веществ отсутствуют, то [К] = СK и [А] = СА, что дает следующее уравнение для скорости реакции
Реакция имеет первые порядки по катализатору и реагенту первой стадии, нулевок порядок по реагенту второй стадии и не тормозится веществом Z. Ее константа скорости равна константе скорости элементарной стадии образования промежуточного комплекса (k1), которую легко найти, разделив экспериментально найденную эффективную константу kэф на концентрацию катализатора.
Следовательно, в данном случае кинетическое уравнение включает характеристики только первой стадии процесса, которая оказывается лимитирующей.
2. Скорость второй стадии реакции значительно меньше скорости обратного превращения промежуточного комплекса в исходные реагенты, т. е. k2 [Y]<< k-1 [Z]. Это позволяет пренебречь величиной k2[Y] в знаменателе и уравнение 14.1 принимает вид:
[ ][ |
][ |
] |
[ ][ |
][ |
] |
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
[ |
] |
|
|
[ |
|
] |
|
|
|
|
|
|
||
В кинетическое уравнение (14.3) входят константа скорости второй стадии и |
||||||||||||||
лишь константа равновесия первой, |
т.е. общая скорость процесса лимитируется |
|||||||||||||
второй стадией ‒ превращением промежуточного комплекса в продукты. |
||||||||||||||
Равновесная концентрация промежуточного комплекса может достичь зна- |
||||||||||||||
чительной величины, |
и тогда аналитическая концентрация катализатора будет |
|||||||||||||
суммой: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
[ ] |
|
[ |
] |
|
[ ][ ] |
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
[ ] |
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||
откуда |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
[ |
] |
|
|
|
|
|
|
|
|
||
|
|
|
|
|
[ |
] |
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
[ |
] |
|
|
|
|
Если реагенты находятся в избытке, можно принять, что [А] ≈ СА, [Z] = СZ и [Y] = Сy, то кинетическое уравнение (14.3) принимает вид:
( )
При очень малой константе равновесия можно пренебречь в знаменателе слагаемым К1СА, что дает:
227

Если первая стадия состоит в присоединении или вещество Z является растворителем и его концентрация входит в выражение для константы, получим:
Оно становится подобным по форме с (14.2), и два разных механизма оказываются кинетически неразличными. Для решения вопроса о лимитирующей стадии приходится прибегать к другим способам, например к определению энтропии активации или реакционной способности, кинетическому изотопному методу и др.
Если константа равновесия при образовании промежуточного комплекса достаточно велика, слагаемым К1СА в знаменателе уравнения уже пренебречь нельзя. В отсутствие вещества Z (при первой стадии присоединения) или при наличии его в большом избытке (растворитель) уравнение дает:
Если Y является растворителем или если вторая стадия мономолекулярна, из уравнения (14.5) выпадает Су. При очень большой константе К1СА иногда можно пренебречь в знаменателе единицей по сравнению с К1СА, в результате чего получим:
Оказывается, что в этом случае реакция имеет нулевой порядок по реагенту А первой стадии. Физический смысл этого состоит в том, что ввиду почти количественного образования промежуточного комплекса дальнейшее повышение концентрации А уже не ведет к ускорению реакции.
Из уравнений видно, что даже для одной и той же реакции может наблюдаться разная зависимость скорости от концентрации реагента А. При малой ее величине, если можно пренебречь К1СА по сравнению с СZ или единицей, имеем линейную зависимость (первый порядок по А). Наоборот, при высокой СА, если можно пренебречь СZ или единицей в знаменателе, получим, что скорость не зависит от концентрации этого реагента (нулевой порядок по А). В промежуточной области концентраций будет наблюдаться постепенно замедляющийся рост скорости.
Эти закономерности изображены на рис. 14.1, причем по горизонтальному участку кривой легко находится истинная константа скорости каталитической реакции (k2), равная эффективной константе, деленной на концентрацию катализатора.
Рассмотренный тип кинетических уравнений со второй лимитирующей стадией и значительной концентрацией промежуточного комплекса также нередко встречается в органическом синтезе.
Так, при взаимодействии α‒окисей с кислотными реагентами (фенолами, карбо-
228

новыми кислотами) и нуклеофильном катализе сопряженными основаниями комплекс α‒окиси с реагентом образуется в значительном количестве, и реакция слабо ускоряется с повышением концентрации фенола или карбоновой кислоты.
Другой пример ‒ эпоксидирование аллилового спирта перекисью водорода в присутствии вольфрамовой кислоты, которая быстро окисляется перекисью в надвольфрамовую кислоту, и весь катализатор практически находится только в этой форме. В результате реакция имеет нулевой порядок по перекиси водорода и первый ‒ по катализатору и аллиловому спирту в соответствии с уравнением
(14.6):
Встречаются и такие реакции, когда катализатор находится в большом избытке, а концентрация реагента, наоборот, незначительна (сульфирование и сульфатирование слабо растворимых в серной кислоте веществ, нитрование смесями серной и азотной кислот и др.). Здесь можно принять, что [К] ≈ Ск и [Y] ≈
Су, но концентрация А определяется выражением: |
|
[ ] [ ] [ ] |
[ ][ ] |
Подставляя находимую из этого уравнения величину [А] в уравнение скорости, получим:
В этом случае имеется переменная зависимость скорости (аналогичная изображенной на рис. 14.1) от концентрации не реагента, а катализатора, причем по горизонтальному участку кривой можно вновь определить истинную константу скорости каталитической реакции.
14.2. Кислотно-основный катализ
К кислотно-основному катализу применимы изложенные выше закономерности, в то же время имеется ряд особенностей.
В современной химии используются две теории кислот и оснований: протонная теория Брёнстеда-Лоури и электронная теория Льюиса.
14.2.1. Кислоты и основания Брёнстеда
По Брёнстеду-Лоури кислотами называют вещества, способные поставлять (отдавать) протон, а основания – вещества, способные присоединять протон:
AH |
|
H |
A |
H |
B: |
|
BH |
Кислота и основание в растворе взаимосвязаны сопряженным процессом переноса протона. В растворе протон не существует в свободном виде и находится только в сольватированном состоянии (например, в воде Н3О+, Н5О2+, Н7О3+). Кислотный характер соединения (АН или ВН+) может проявляться только в присутствии основания (В:, А–) или растворителя (SН), обладающего основными свойствами, и наоборот, основный характер соединения (В:, А–)
229



