

2. Гликозилирование ароматических соединений
Первый синтез фенилглюкозида 51в основных условиях осуществлен американским химиком А. Михаэлем в 1879 г. взаимодействием полного ацетата α-D-глюкопиранозилхлорида 49с фенолятом натрия 50 в 96% этаноле (схема 11) [5]. Использование фенолят-анионами, в качестве нуклеофильных агентов для полученияО-β-арилгликозидов сопровождается частичным дезацетилированием как исходных гликозил-доноров, так и целевых продуктов реакции, гидролизом связи С-1 – галоген, что обусловливает низкие выходы получения соответствующих гликозидов. Высокая нуклеофильность фенолят-иона позволяет проводить эту реакцию с высоким выходом и в мягких условиях, причем гидролиз ацилгалогеноз не успевает пройти в заметной степени и не может служить серьезной помехой основной реакции. Конденсация, как правило, приводит к 1,2-транс-гликозидам.
Схема 11

Для синтеза арилгликозидов применялись в основном два метода. Первый основан на сплавлении полных ацетатов сахаров с фенолами в присутствии различных кислотных катализаторов [22]. Второй заключается во взаимодействии фенолятов щелочных металлов с ацилгликозилбромидами или ацилгликозилхлоридами в водном ацетоне или смеси спирт-хлороформ [23]. Известны также многочисленные модификации метода Гельфериха [7].
Ни один из указанных методов не является универсальным. Наиболее общий из них, метод сплавления, применим лишь для низкоплавких монофункциональных фенолов. Второй по значению метод, основанный на использовании фенолятов, не пригоден для синтеза арилгликозидов с электронодонорными заместителями в ядре [23], а также для синтеза производных некоторых сахаров, например рамнозы [24]. Модификации метода Гельфериха не могут быть использованы в случае полифункциональных фенолов [25]. Таким образом, известные методы синтеза страдают серьезными недостатками при гликозилировании полифункциональных фенолов, несущих электронодонорные заместители, а также при распространении их на некоторые классы углеводов, типа пентоз или дезоксигексоз.
Развитие методов межфазного катализа в этой области биоорганической химии позволило получить малодоступные другими методами производные, существенным образом повысить селективность ряда процессов и значительно увеличить выходы продуктов многих химических превращений моносахаридов.
Авторами работы [3] на примере N-ацетилированных 4‑нитрофенилгликозидов была показано возможность получения соединений, потенциально являющихся хромогенными субстратами для изучения гиалуронидазы. Гликозилирование проводилось в условиях межфазного катализа в системе «метиленхлорид – 1М раствор карбонат калия» с применением в качестве катализатора тетрабутиламмония гидросульфата (ТБАГ). Замена 1-О-ацильного производного на метилгликозид гиалуроновой кислоты приводило к невысоким выходам 4-нитрофенилглюкозида. Причиной этого является низкая концентрация образующего в реакционной системе α-бромида гиалуроновой кислоты.
Схема12

Для качественной и количественной оценки работы ферментов, рассматриваются объекты, как природного происхождения, так и синтетические. Например, агликоноподобные α-кетозидазы должны обеспечивать легкое их обнаружение с помощью спектроскопических свойств. Арильные производные N-ацетилнейраминовой кислоты оказались удобными для данных исследований. Ряд методов для стереоселективного получения α-кетозидов из N‑ацетилнейраминовой кислоты, широко описаны в литературе и содержат ряд недостатков. Например, по методу Кенигса-Kнoрра получение подобных производных требовало применение дорогих катализаторов, предусматривало многостадийный процесс или продолжительный синтез, протекающий зачастую с низкими выходами. Одной из причин невысоких выходов являлась низкая растворимость образующихся солей ароматических соединений в органических растворителях. Проведение реакции в межфазных условиях позволяла исключить эти недостатки. Так, Джордж Ротермель с сотрудниками показал [26], что гликозилирование N‑ацетилнейраминовой кислоты в условиях межфазного катализа в системе «хлороформ-водного раствора щелочи» с применением в качестве катализатора бензилтриэтиламмоний хлорида (ТЭБАХ) позволило получить известные и новые арил-α-кетозиды с высокой стереоселективностью. Реакция протекала быстро и приводила к хорошим выходам арилпроизводных. Например, 4-метилумбеллиферил α-кетозид, являющийся стандартным субстратом нейраминидаз, в данных условиях получен с выходом 70%. Для получения 4-метилумбеллифирильного производного необходимо использование 0,1 н. раствора основания. Уменьшение и увеличение концентрации раствора щелочи приводило к протеканию побочных процессов: гидролиза гликозил-галогенида; деструкции 4-метилумбеллиферона. Авторам работы удалось синтезировать новый кетозид флуоресцеин N‑ацетилнейраминовой кислоты. Монокетозид получен с выходом 40%, тогда как метод Кенигса-Kнoрра приводил к получению гликозида с выходом ниже 10%. В условиях межфазного катализа идентифицировано образование в небольших количествах побочного продукта – биспроизводного N‑ацетилнейраминовой кислоты, выделение которого не представлял затруднений, что еще раз показывает преимущество данного подхода, в отличие от метода Кенигса-Kнoрра.
Схема 13
Взаимодействием ацетобромглюкозы в условиях межфазного катализа получен п‑формилглюкозид с выходом 55%. Реакция проводилась в системе хлороформ – водный раствор карбоната калия в присутствии тетрабутиламмоний бромида [26].
Схема14
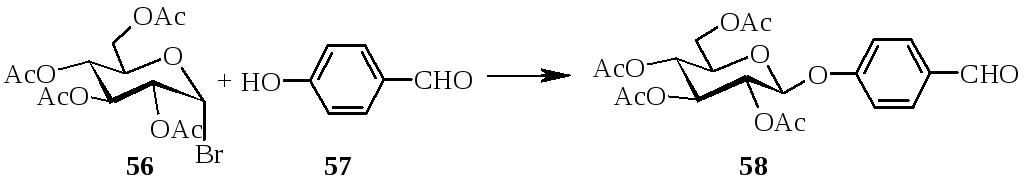
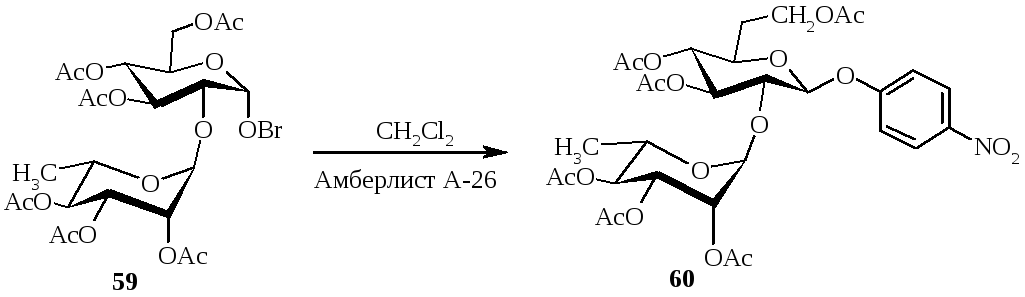
Авторами работы [4] было показано, что использование р-нитрофенил-2-О-α-L-фукопиранозил-β-D-галактопиранозида в качестве субстрата позволяло обнаружить α-L-фукозидазу, имеющую специфичность к α-(1→2)-связи. р-Нитрофенил-3,4,6-три-O-ацетил-β-D-галактопиранозид получен в условиях межфазного катализа. Реакция 3,4,6-три-О-ацетил-2-O-(2,3,4-три-О-ацетил-α-L-фукопиранозил)-α-D-галактопиранозилбромида с р-нитрофеноксидом протекала в среде метиленхлорида в присутствии амберлиста А-26 с выходом продукта реакции 34%.
Схема15
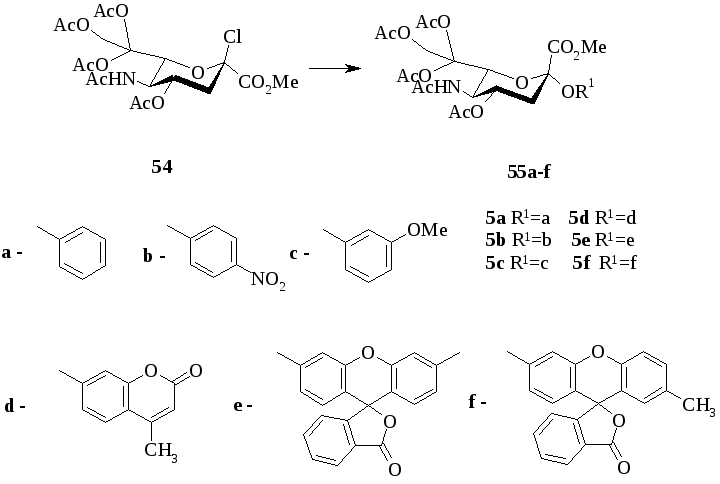
В работе [5] авторы показали возможность получение 4-гидроксихинолин-β-гликозида. Соединения хинина известны как противомалярийные препараты. Анализ синтетических аналогов, обладающих противомалярийным действием, в настоящее время остается актуальным.
Схема16

Замещенные фенилгликозиды широко используется для обнаружения ферментативного расщепления гликозидной связи. В работе [5] был получен ряд замещенных фенил-2-ацетамидо-2-дезокси-α-и β‑D‑глюкопиранозидов. Замещенные фенил-2-ацетамидо-2-дезокси-β-D-глюкопиранозиды были получены реакцией α-хлорида N‑ацетилглюкозамина с фенолятом натрия по методу Кенигса-Кнора. Взаимодействием сполна ацетилированных глюкозамина с различными фенолом в присутствии безводного хлорида цинка получены соответствующие α-гликозиды (схема17). Выходы α- и β-D-глюкопиранозидов приведены в таблице 3.
Схема17
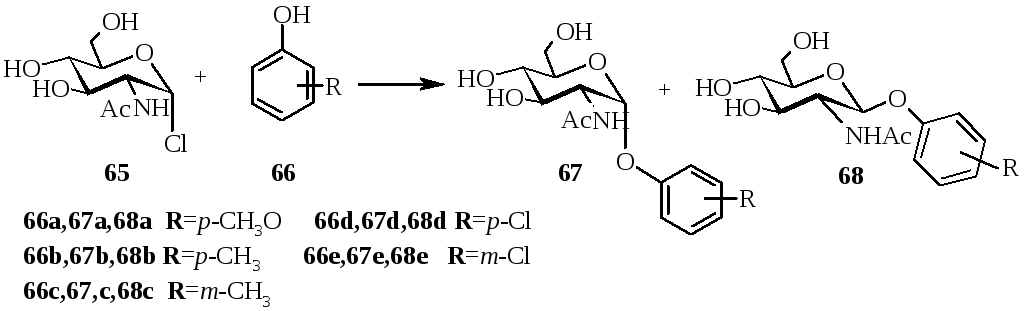
Таблица 3.
Выходы замещенных фенил 2-ацетамидо-2-дезокси-D-глюкопиранозидов
|
Заместители |
Формула |
Выход |
Заместители |
Формула |
Выход |
|
Фенил-3,4,6-три-O-ацетил-2-ацетомидо-2-диокси-α-D-глюкопиранозиды |
Фенил-3,4,6-три-O-ацетил-2-ацетомидо-2-диокси-β-D-глюкопиранозиды | ||||
|
p-CH3O |
C21H27O10N |
17 |
p-CH3O |
C21H27O10N |
13 |
|
p-CH3 |
C21H27O9N |
30 |
p-CH3 |
C21H27O9N |
27 |
|
m-CH3 |
C21H27O9N |
24 |
m-CH3 |
C21H27O9N |
38 |
|
p-Cl |
C20H24O9NCl |
17 |
p-Cl |
C20H24O9NCl |
8 |
|
m-Cl |
C20H24O9NCl |
16 |
m-Cl |
C20H24O9NCl |
11 |
Ранее авторами работы синтезирован ряд гликозидов трет-бутил-4-гидрохинона с целью изучения их антиоксидантных свойств. Подобные соединения ингибируют липидное окисление. В качестве исходных гликозил-доноров рассматривались сполна ацетилированные D-глюкоза, D-галактоза и 2-дезокси-2-бутанамидоглюкопиранозилхлорид. Были изучены условия гликозилирования трет-бутил-4-гидрохинона (табл. 4). Во всех случаях реакция протекала с образованием смеси аномеров. Суммарный выход α- и β-гликозидов составил от 33 до 40%. Образование продуктов с β‑конфигурации гликозидной связи наблюдалось в большей степени (табл. 4). В качестве побочного продукта идентифицирован гликозид 72 с выходом 2% (схема 18). Использование трифлата иттербия (Yb(OTf)3) уменьшало время реакции, но не влияет на выход продуктов и стереоселективность реакции. Взаимодействием пентаацетата β-D-галактозы 70b с замещенным гидрохиноном получен соответствующий β-галактозид с выходом 40% (табл. 4). Введение 2-дезоксисахара в молекулу двухатомного фенола стало возможным при использовании β-D-глюкопиранозилхлорида. 2-Аминогликозид гидрохинона получен с выходом 60%.
Схема18

Таблица 4.
Выходы гликозидов трет-бутилгидрохинона
|
Донор |
Условия |
Продукт |
Суммарный выход,(%) |
Соотношение α/β |
|
69a |
(p-СH3)PhSO3H,125оС |
71a/71a+72 |
33+2 |
29/71 |
|
69a |
(p-СH3)PhSO3H, CH2Cl2 |
71a/71a |
67 |
4/96 |
|
69a |
(p-СH3)PhSO3H, Yb(OTf)3, CH2Cl2 |
71a |
61 |
10/90 |
|
70b |
(p-СH3)PhSO3H, CH2Cl2 |
71b/71b |
40 |
15/85 |
|
70c |
ZnCl2, CH2Cl2 |
71c/71c |
60 |
11/89 |
|
70b |
ZnCl2, CH2Cl2 |
71d/71d |
40 |
5/95 |