

pCmin < lg ' – 6. |
(4.42) |
Но эта формула в ряде случаев (особенно при образовании прочных комплексов) дает неверные, слишком оптимистичные прогнозы. Формула (4.42) не учитывает многих факторов, влияющих на нижнюю границу определяемых содержаний (скорость реакций, наличие примесей в реагентах и растворителе, индикаторные погрешности). На практике комплексонометрическое титрование с металлохромными индикаторами дает достаточную точность лишь при концентрации больше 10–4 М. Использование инструментальных методов для выявления точки эквивалентности позволяет снизить Сmin еще на 1–2 порядка.
4.6. Осадительное титрование. Аргентометрия
Процессы осаждения отвечают требованиям титриметрического анализа в меньшей степени, чем реакции комплексообразования. Осложнения здесь могут возникнуть не только из-за нестехиометрического или неколичественного протекания реакций, но и вследствие соосаждения. Важно и то, что реакции осаждения сопровождаются образованием пересыщенных растворов, равновесие устанавливается слишком медленно. Поэтому осадительное титрование в контрольноаналитических лабораториях применяют реже, чем методы нейтрализации или комплексометрии. Поскольку скачок на кривой осадительного титрования выражен тем больше, чем меньше растворимость образующегося осадка, для титрования можно использовать лишь реакции и условия, ведущие к образованию наименее растворимых соединений. Это галогениды серебра, сульфаты бария и свинца, фосфаты и гексацианоферраты ряда металлов, а также соединения, образованные органическими осадителями (оксихинолин и др.). Из всех осадительных методов наибольшее значение имеет аргентометрия, созданная в 30-х гг. XIX века знаменитым французским химиком Ж. Гей-Люссаком для определения серебра. Позднее нитрат серебра стали применять в качестве титранта при определении галогенидов и некоторых других анионов. Используют и другие титранты – Hg2(NO3)2 (меркурометрия), K4[Fe(CN)6] (гексацианоферратометрия), BaCl2 и Н2SO4 (сульфатометрия).
С появлением комплексонометрии (гораздо более надежного и чувствительного, хотя и менее селективного метода) осадительное титрование почти перестали применять для определения металлов, но метод сохранил свое значение при определении неметаллов и ор-
281

ганических веществ. Способы контроля к.т.т. в осадительном титровании весьма разнообразны, мы рассмотрим их далее на примере метода аргентометрии.
Аргентометрическое титрование. Титрантом служит раствор
AgNO3, обычно 0,1–0,01 М. Определяемые вещества – хлорид, бромид, иодид, тиоцианат (роданид) и некоторые другие анионы. Аргентометрическое титрование однозарядных анионов Х– соответствует схеме: Х– + Ag+ AgХ ↓. Аргентометрическим методом анализируют химические реактивы, природные и сточные воды, пищевые продукты, лекарственные препараты и др. С помощью того же титранта по методу замещения определяют галогенсодержащие органические соединения. Известны также аргентометрические методы определения карбоновых кислот, а также некоторых фосфор- и серосодержащих органических веществ.
Нитрат серебра в растворе может восстанавливаться примесями органических веществ, а также подвергаться фотохимическому разложению. Поэтому приготовленный раствор AgNO3 стандартизуют по точной навеске предварительно перекристаллизованного и высушенного хлорида натрия. Стандартизованный раствор AgNO3 хранят в склянке темного стекла в защищенном от света месте.
Способы обнаружения конечной точки титрования. В ар-
гентометрии, как и в осадительном титровании в целом, используют индикаторы разного типа, в частности, осадительные, металлохромные и адсорбционные. Визуальные методы контроля к.т.т. в аргентометрии назвали по фамилиям первооткрывателей.
Аргентометрическое титрование по методу Мора основано на применении осадительного индикатора хромата калия, образующего вблизи т.экв. красный осадок хромата серебра:
2Ag++ СrO42– Ag2СrO4тв .
В этом методе важен правильный выбор концентрации индикатора: изменение цвета осадка должно происходить в пределах скачка титрования, как можно ближе к т.экв. Например, при титрова-
нии хлоридов значение в т.экв. [Ag+] = 
 ПРAgCl = 1,3·10–5 М. Если
ПРAgCl = 1,3·10–5 М. Если
считать, что именно в этот момент должно начаться образование индикаторного осадка Ag2СrO4, можно найти, какую концентрацию индикатора (хромат-ионов) следует создавать в ходе титрования:
[СrO42–] = ПРAg2СrO4 / [Ag+]2 = 1,1·10–12/(1,3·10–5)2 = 6,3·10–3 моль/л.
282
На практике готовят 5 %-й раствор индикатора K2СrO4 и добавляют его к анализируемому раствору до концентрации ≈ 0,005 М. По методу Мора определяют прямым титрованием только хлорид и бромид при рН 6,5–10,3. В кислых средах образованию Ag2СrO4 препятствует протолиз индикатора:
СrO42– + Н3O+ НСrO4– + Н2O, 2НСrO4– Сr2O72– + Н2O,
а в щелочных средах титрант разлагается с образованием коричневого осадка Ag2O:
2Ag+ + 2OН– = 2AgOН ↓ = Ag2O ↓ + Н2O.
Требуемое значение рН обычно поддерживают добавлением
NaHCO3.
Метод Фаянса основан на применении адсорбционных индикаторов (флуоресцеин, эозин и др.). Адсорбционные индикаторы представляют собой слабые органические кислоты. Анионы индикатора в растворе и в адсорбированном состоянии на поверхности осадка имеют различную окраску (так, флуоресцеин в растворе жел- то-зеленый, а в адсорбированном состоянии придает осадку розовый цвет). Анионы – хлорид, бромид, иодид, цианид, тиоцианат, селенит
– по методу Фаянса определяют прямым титрованием стандартным раствором нитрата серебра. До т.экв. поверхность осадка AgХ заряжена отрицательно вследствие адсорбции анионов Х-, находящихся в растворе в избытке. Отрицательный заряд поверхности осадка препятствует адсорбции анионов индикатора, осадок остается неокрашенным. После т.экв. добавление лишней капли титранта приводит к адсорбции на поверхности осадка катионов Ag+, происходит перезарядка поверхности. Теперь на положительно заряженной поверхности осадка адсорбируются анионы индикатора, поверхностный адсорбционный комплекс этих анионов с катионами серебра придает осадку окраску – таким образом фиксируется к.т.т. Выбор среды для титрования определяется кислотной константой индикатора, его способностью образовать в растворе достаточное количество анионов: с более слабой кислотой флуоресцеином титруют при рН 6,5–10,3, с индикаторами – более сильными кислотами (эозин и др.) – при рН 2–10,3.
В аргентометрическом методе Фольгарда в качестве титранта для ионов Ag+ используют стандартный раствор тиоцианата (роданида) калия, в качестве индикатора – раствор железоаммонийных квасцов NH4Fe(SO4)2 · 12Н2O (~0,01 М); ион Fe3+ образует красный тиоцианатный комплекс при добавлении лишней капли титранта:
283
Ag+ + SCN– AgSCN ↓ |
белый |
Fe3+ + n SCN– [Fe(SCN)n](3–n)+ |
красный |
Прямым титрованием по методу Фольгарда определяют катионы серебра, обратным титрованием (титруя тиоцианатом непрореагировавший остаток AgNO3) – многие анионы: хлорид, бромид, иодид, тиоцианат, карбонат, фосфат, сульфид, хромат, цианид, оксалат, арсенат и др. При определении хлоридов возможен перерасход тиоцианата, связанный с частичным переходом осадка АgCl в менее растворимый AgSCN: AgСl ↓ + SCN– AgSCN ↓ + Сl–. Поэтому перед титрованием необходимо первоначально образовавшийся осадок AgCl отфильтровать или изолировать от раствора добавлением тяжелого несмешивающегося с водой растворителя (нитробензола). Титрование по методу Фольгарда проводят в кислых средах, иначе индикатор неприменим (осаждается в виде Fe(ОН)3).
Все визуальные методы аргентометрии не очень чувствительны и мало селективны. При титровании разбавленных растворов или смесей чаще применяют инструментальные методы обнаружения к.т.т., например, потенциометрию с индикаторным серебряным электродом, потенциал которого линейно зависит от величины pAg.
Кривые аргентометрического титрования*. Кривые титрования в аргентометрии строят в координатах pAg – f (или рХ – f). Для расчета pAg в разные моменты титрования используют ту же упрощенную модель, что и в других вариантах титриметрии (не учитывают разбавление раствора, ионную силу, скорость установления равновесия и т. п.). В рамках этой модели можно вывести расчетные формулы для прогнозирования величины pAg (см. табл. 4.18). Способ их вывода аналогичен тем, которые были использованы в разделах 4.4 и 4.5.2. Если ионы Ag+ или Х– участвуют в побочных реакциях, вместо концентрационного ПР используют условное, которое находят с учетом поправок на маскирование, аналогичных поправкам p Y и p М в комплексонометрии. Обычно, однако, влияние побочных процессов на аргентометрическое титрование незначительно. По формулам, приведенным в табл. 4.18, можно построить кривые титрования однокомпонентных модельных растворов разных галогенидов (рис. 4.17). Для определения границ скачка примем допустимую погрешность равной 1 %. Тогда:
∆ pAg 1% = pAg 0,99 – pAg 1,01 = pПР – 2pС – 4. |
(4.43) |
284
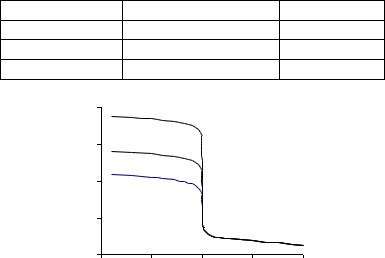
Таблица 4.19
Формулы для расчета кривых аргентометрического титрования
Стадия титрования |
Степень оттитрованности |
рAg |
||
до ТЭ |
|
0 < f < 1 |
|
pПР – pС + lg(1–f) |
в ТЭ |
|
f = 1 |
|
½ pПР |
после ТЭ |
|
f > 1 |
|
pС– lg(f –1) |
pAg 16 |
|
|
|
|
|
I– |
|
|
|
12 |
|
|
|
|
|
Br– |
|
|
|
pAg 8 |
Cl– |
|
|
|
4 |
|
|
|
|
0 |
|
|
|
f |
|
|
|
|
|
0 |
0,5 |
1 |
1,5 |
2 |
Рис. 4.14. Кривые аргентометрическогоf титрования 0,1 М растворов Сl–, Вr– и I–
Чем менее растворим образующийся осадок (выше рПР) и чем выше концентрация титруемого раствора, тем больше высота скачка и, следовательно, тем выше точность титрования. В аргентометрии следует избе-
гать повышения температуры и присутствия веществ, образующих с ионами серебра растворимые комплексные соединения. Для понижения ПР нередко осадительное титрование ведут в смешанных водно-органических средах. Так, в 50 %-м этаноле для осадка AgСl pПР = 10,93 (в воде 9,75).
Раздельное аргентометрическое определение нескольких анионов в смеси удается только при значительном различии ПР образующихся осадков. Так, теоретически возможно последовательное титрование трех галоге- нид-ионов раствором AgNO3: вначале осаждается иодид, дающий наименее растворимый осадок, затем бромид и хлорид. На практике при таком титровании возможны систематические погрешности, связанные с адсорбцией и соосаждением ионов.
285
4.7. Окислительно-восстановительное титрование (редоксметрические методы)
Еще в конце XVIII века в объемном анализе стали использовать реакции, которые теперь принято называть окислительновосстановительными. С тех пор было разработано довольно много разных редоксметрических методов, и все они основаны на одном и том же принципе: восстановители титруют стандартными растворами окислителей и наоборот. Но растворы восстановителей могут окисляться кислородом воздуха (раздел 3.6), их необходимо стандартизовать непосредственно перед применением. Поэтому методы, основанные на применении титрантов-восстановителей (титанометрия, аскорбинометрия и др.), менее распространены. В качестве титрантов обычно используют более устойчивые растворы окислителей (перманганатометрия, хроматометрия, иодометрия, цериметрия, броматометрия, ванадатометрия и др.).
4.7.1. Кривые редоксметрического титрования. Редокс-индикаторы
Запишем реакцию титрования, а также обе ее составляющие (полуреакции):
z2Red1 + z1Ox2 z2Ox1 + z1Red2 Red1 – z1e– Ox1
Ox2 + z2e– Red2
Исходную молярную концентрацию титруемого вещества Red1 обозначим символом С. Рассчитаем кривую титрования, как зависимость равновесного редокс-потенциала (Е) от степени оттитрованности (f). Для расчета используем уравнение Нернста. В условиях равновесия потенциал можно рассчитывать по любой из полуреакций (Е1 = Е2). Удобнее до т.экв. вести расчет по первой полуреакции, отвечающей титруемому веществу, а за т.экв. – по второй полуреакции.
Е1 = E10 + |
0 ,059 |
lg |
[Ox1 |
] |
|
, |
(4.44) |
|||
z1 |
|
[Red1 ] |
|
|||||||
|
|
|
|
|
||||||
Е2 = E20 + |
|
0 ,059 |
lg |
[Ox 2 |
] |
|
. |
(4.45) |
||
|
z 2 |
|
[Red 2 ] |
|||||||
|
|
|
|
|
||||||
В начальный момент титрования расчет равновесного потенциала невозможен, так как в растворе присутствует лишь одно веще-
286

ство (восстановленная форма Red1). До т.экв. концентрация восстановленной формы определяемого вещества уменьшается: [Red1] = С(1 – f), а окисленной формы растет: [Ox1] = С f. Подставляя эти выражения в формулу (4.44), получаем:
|
Е = E 0 |
+ |
0 ,059 |
lg |
f |
. |
(4.46) |
|
1 |
|
z1 |
1 f |
|
||
|
|
|
|
||||
Из |
(4.46) следует, что наполовину оттитрованный |
раствор |
|||||
(f = 0,5) |
характеризуется равновесным |
потенциалом, равным стан- |
|||||
дартному потенциалу титруемой редокс-пары.
Теперь выведем формулу для расчета Е в т.экв.1 Для этого умножим уравнение (4.44) на z1, (4.45) на z2 , а затем сложим получен-
ные выражения: |
|
|
|
|
|
|
(z1+ z2)Ет.экв. = z1 E10 + z2 E20 |
+ 0,059 lg |
[Ox1 |
][Ox2 |
] |
. |
|
[Red1 |
][Red |
2 ] |
||||
|
|
|
Непрореагировавшие остатки веществ Red1 и Ox2 в т.экв. находятся в растворе в количествах, эквивалентных друг другу. Отношение их концентраций равно отношению стехиометрических коэффи-
циентов: |
[Ox 2 ] |
|
z1 |
|
. Для продуктов выполняется аналогичное со- |
||||||
|
|
|
|
||||||||
[Red1 ] |
z 2 |
|
|
|
|
|
|
||||
отношение: |
[Ox1 ] |
|
|
z 2 |
. Поэтому lg |
[Ox1 |
][Ox2 ] |
= lg 1 = 0. Полу- |
|||
|
|
|
|
|
|
||||||
|
[Red 2 ] |
|
z1 |
[Red1 |
][Red2 ] |
|
|||||
чаем важную расчетную формулу: |
|
|
|
||||||||
|
z E0 |
z |
2 |
E0 |
|
|
|
|||
Ет.экв = |
1 |
1 |
|
|
2 |
|
. |
(4.47) |
||
|
z1 |
z2 |
|
|
|
|||||
|
|
|
|
|
|
|
||||
Для тех реакций, где z1 = z2: |
|
|
|
|
|
|
|
|
||
|
|
E0 |
E0 |
|
|
|
|
|
||
Ет.экв = |
1 |
|
2 |
|
|
. |
|
(4.47а) |
||
|
2 |
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
Нередко титрование ведут в условиях, отличающихся от стандартных. Поскольку на величину равновесного потенциала многих
1 Формулы (4.45–4.48), собранные в табл. 4.20, не предназначены для запоминания. При решении расчетных задач их без особого труда можно самостоятельно вывести из уравнения Нернста.
287
полуреакций влияют рН раствора и реакции комплексообразования, вместо стандартных потенциалов следует использовать формальные (реальные) потенциалы полуреакций (значения Е0 ), вычисленные, как описано в разделе 3.6.
После т.экв. в растворе растет концентрация избыточного титранта. Концентрация продукта Red2 при этом остается неизменной, она определяется исходной концентрацией титруемого вещества и стехиометрическими коэффициентами. После подстановки в (4.45) получаем:
[Ox2] = |
z1 |
|
С(f – 1); |
[Red2] = |
z1 |
С. |
|||
z 2 |
|
z 2 |
|||||||
|
|
|
|
|
|
|
|
||
Е = |
E 0 |
+ |
0 ,059 |
|
lg(f – 1). |
|
(4.48) |
||
|
|
||||||||
|
|
2 |
|
z 2 |
|
|
|
||
|
|
|
|
|
|
|
|
||
Выведенные формулы собраны в табл. 4.20. Воспользуемся ими, чтобы построить кривые перманганатометрического титрования двух восстановителей – железа(II) и пероксида водорода в стандартных условиях (рН = 0).
Реакция, идущая в ходе первого титрования:
5 Fe2+ + MnO4– + 8 H+ = 5 Fe3+ + Mn2+ + 4 H2O.
Реакция, идущая в ходе второго титрования:
5 Sn2+ + 2 MnO4– + 16H+ = 5 Sn4+ + 2 Mn2+ + 8 H2O.
Таблица 4.20
Формулы для расчета кривых редоксметрического титрования
Стадия |
Степень |
|
|
|
|
|
|
Е |
|
|
|
|
|
|
титрования |
оттитрованности |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
до т.экв. |
0 < f < 1 |
E10 + |
0 ,059 |
lg |
f |
|
||||||||
|
|
1 f |
||||||||||||
|
|
|
|
|
|
|
|
z1 |
|
|
|
|||
|
|
|
z |
1 |
E 0 |
z |
2 |
E 0 |
||||||
в т.экв. |
f = 1 |
|
|
|
1 |
|
|
2 |
|
|
||||
|
|
|
|
|
z1 z 2 |
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|||||||||
после т.экв. |
f > 1 |
E20 + |
|
0 ,059 |
lg(f – 1) |
|||||||||
|
|
|||||||||||||
|
|
|
|
|
|
|
|
z 2 |
|
|
|
|
|
|
Для редокс-пар Fe3+/Fe2+, Sn4+/Sn2+ и MnO4–/ Mn2+ стандартные потенциалы, соответственно, равны 0,77 В, 0,15 В и 1,51 В. Результа-
288
ты подстановки приведены в табл. 4.21, а сами кривые показаны на рис. 4.15.
Таблица 4.21
Данные для построения кривых редоксметрического титрования
f |
|
|
|
Расчетная |
Значения E в ходе реакции |
|||||||||||||
|
|
|
формула |
|
|
|
Окисление Fe(II) |
Окисление Sn(II) |
||||||||||
|
|
|
|
|
|
|
||||||||||||
0,5 |
|
|
|
|
E |
0 |
|
|
|
|
|
|
|
0,77 |
0,15 |
|||
|
|
|
|
|
|
|
1 |
|
|
|
|
|
|
|
|
|
||
0,9 |
|
|
|
E10 |
+ |
|
0,059 |
|
|
0,83 |
0,18 |
|||||||
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
z1 |
|
|
|
|
|
|||
0,99 |
|
|
|
E10 |
+ |
0 ,12 |
|
|
|
|
0,89 |
0,21 |
||||||
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
z1 |
|
|
|
|
|
|||
|
|
z |
1 |
E 0 |
z |
2 |
E |
0 |
|
|
|
|||||||
1 |
|
|
1 |
|
|
|
|
|
|
|
|
2 |
|
1,31 |
1,11 |
|||
|
|
|
z1 z 2 |
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
1,01 |
|
|
|
E20 – |
0 ,12 |
|
|
|
|
1,49 |
1,49 |
|||||||
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
z 2 |
|
|
|
|
|
|
|
|
|
1,1 |
|
|
|
E20 – |
0 ,059 |
|
1,50 |
1,50 |
||||||||||
|
|
|
|
|||||||||||||||
|
|
|
|
|
|
|
|
|
z 2 |
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
E |
0 |
|
|
|
|
|
|
|
1,51 |
1,51 |
|||
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
||||
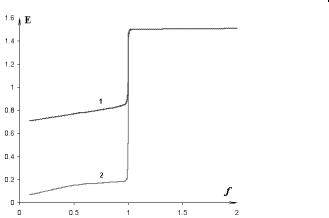
Рис. 4.15. Перманганатометрическое титрование двух восстановителей разной силы: 1 – ионы железа(II); 2 – ионы олова(II)
289

Кривые редоксметрического титрования могут быть несимметричны. Если для полуреакций z1≠ z2, т.экв. находится не посередине скачка (в рассматриваемом примере – ближе к его верхней границе). Для погрешности ± 1 % высота скачка титрования теоретически равна:
∆ Е±1% = Е1,01 – Е0,99 |
≈ ( E |
0 |
– |
0,12 |
|
) – ( E 0 |
+ |
0,12 |
) = |
|||||
|
|
|||||||||||||
|
|
|
|
2 |
|
|
z2 |
1 |
|
z1 |
||||
|
|
|
|
|
|
|
|
|
|
|
|
|||
= E0 |
E0 |
– ( |
0,12 |
+ |
|
0,12 |
). |
(4.49) |
||||||
|
|
|||||||||||||
2 |
|
1 |
|
|
|
z2 |
|
|
z1 |
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
||||
Реальная высота скачка на кривой титрования несколько меньше из-за необратимости редокс-системы окислителя. Но характер изменений высоты скачка выведенная формула показывает правильно. А именно, из (4.49) следует:
1)высота скачка тем больше, чем выше разность стандартных потенциалов окислителя и восстановителя. Это видно и из рис. 4.15:
вслучае олова(II) скачок выше, чем для железа(II);
2)на высоту скачка влияет число электронов, участвующих в полуреакциях. Высота скачка меньше в том случае, когда z1 = z2 = 1;
3)высота скачка редоксметрического титрования, в отличие от
других вариантов титриметрии, не зависит от начальной концентрации титруемого раствора1. Поэтому возможно титрование более разбавленных растворов (иногда удается титровать даже 10–6 М растворы). Однако при титровании растворов с очень низкими концентрациями трудно установить положение к.т.т., да и скорость реакции зачастую недостаточна для титрования.
Ширина интервала перехода типичного редокс-индикатора примерно равна 0,12 В. Это значит, что высота скачка на кривой тит-
рования должна быть не менее 0,12 В. Решение неравенства
E0 |
E0 |
|
0,12 |
|
|
0,12 |
≥ 0,12 позволяет получить критерий воз- |
|
|
||||||
2 |
1 |
|
z2 |
|
z1 |
||
|
|
|
|
||||
можности «одноэлектронного» редоксметрического титрования с погрешностью, не превышающей 1 %:
E 0 |
E 0 |
> 0,36 В. |
(4.50) |
2 |
1 |
|
|
1 Если число частиц окисленной и восстановленной форм в полуреакции различно (например, Cr2O72– / 2Cr3+), формула (4.49) неверна, а высота скачка зависит от концентрации.
290