

МЕХАНИЗМЫ МЕТОДИЧКА
.pdf3.2.3. Реакция сульфирования
Аналогично нитрованию протекает реакция сульфирования бензола с получением сульфокислот. В молекулу ароматического углеводорода можно последовательно ввести одну, две или три сульфогруппы. Монобензолсульфокислота получается при действии на бензол концентрированной серной кислоты на холоду:
С6H6 + HO – SO2OH С6H5 – SO2OH + H2O
Сульфирование проходит по обычному механизму электрофильного замещения. Активным электрофильным агентом является бисульфониевый ион, SO3H+, образующийся в результате протекания реакции
3H2SO4 SO3H+ + H3O+ + 2HSO4-
Далее протекает реакция по известной уже схеме – последовательное образование - и -комплексов и, наконец, продуктов реакции:
Еще более активным электрофильным агентом является оксид серы (VI), в молекуле которого электронная плотность распределена таким образом, что атом серы несет положительный заряд.
Гомологи бензола сульфируются в орто- и пара-положение. При сульфировании ароматических углеводородов дымящей серной кислотой при нагревании и в присутствии катализаторов (Ag2SO4 и др.) получают ди- и три-сульфокислоты. Сульфогруппа при электрофильном замещении ориентирует новый заместитель в мета-положение.
3.2.4. Реакция алкилирования
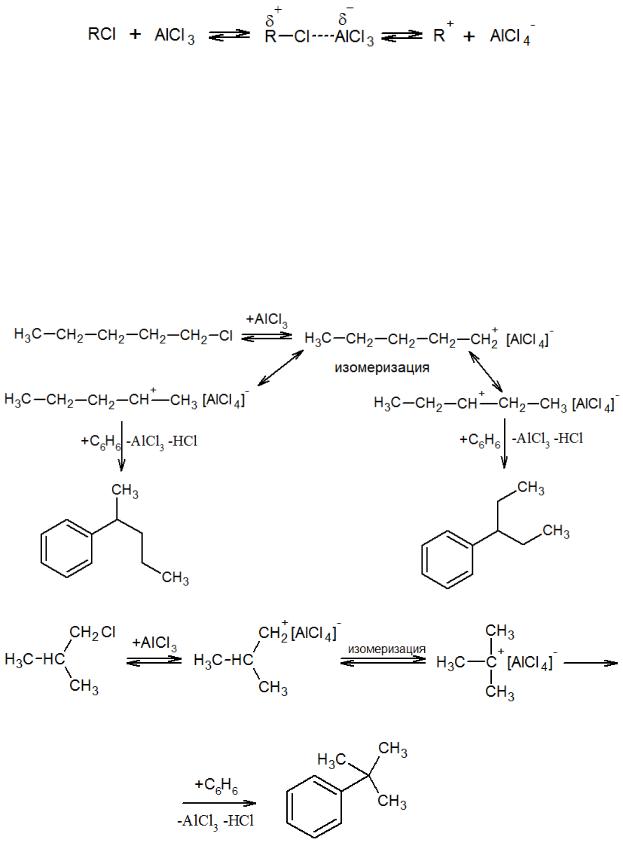
Алкилирование – введение алкильной группы в молекулу органического соединения. Такие реакции носят название реакции Фриделя-Крафтса.
Основными алкилирующими агентами для ароматических соединений являются алкилгалогениды и олефины. В первом случае катализаторами служат кислоты
81
Льюиса, которые, как уже обсуждалось выше, способны поляризовать молекулу реагента, а в пределе – образовать ионные пары:
Степень поляризации будет зависеть от строения алкильной группы: усиление положительного заряда наблюдается с увеличением ее разветвленности, что обусловлено стабилизацией заряда с помощью эффектов гиперконъюгации. А также влияет природа галогена: поляризации способствует увеличение радиуса иона (F < Cl < Br). Как следствие, направление протекания реакции алкилирования определяется устойчивостью образующегося карбкатиона, поэтому, как правило, в реакционных смесях преобладают продукты разветвленной структуры, а именно вторичные и третичные алкилпроизводные бензола. То есть данные процессы также подчиняются правилу Марковникова.
82
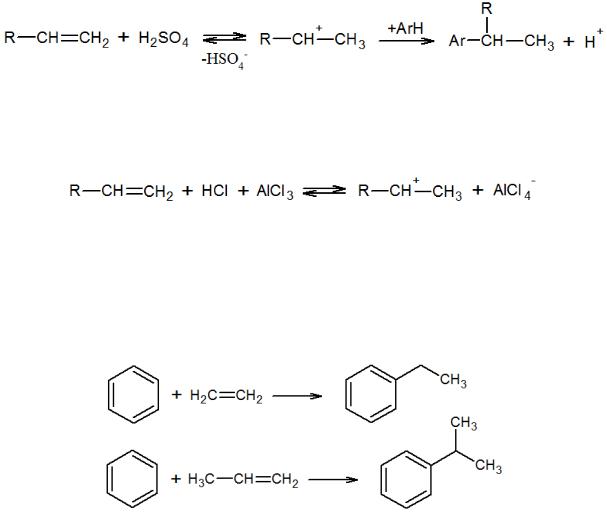
Алкилирование олефинами можно проводить при катализе протонными кислотами, роль которых состоит в промежуточном образовании карбкатиона:
Но чаще всего при алкилировании олефинами используют также кислоты Льюиса, и в этом случае используют сокатализаторы (HCl, RCl, H2O), без которых невозможно образование ионов карбония:
Алкилированием бензола этиленом и пропиленом в промышленности получают такие важные продукты, как этилбензол и кумол. В качестве катализатора используют так называемый комлекс Густавсона, представляющий собой соединение из молекул AlCl3, HCl и 1–6 молекул соответствующего ароматического углеводорода.
3.2.5. |
Реакционная |
способность |
и |
направление |
реакций |
электрофильного замещения в ароматических соединениях
Основная стадия электрофильного замещения атома водорода в ароматическом ядре, которая определяет место будущего заместителя, соответствует не образованию -комплекса, а его перегруппировке в -комплекс. Можно полагать, что переходное состояние этого превращения будет близко по строению к σ- комплексу, при этом заместитель должен ориентироваться в положение с максимально возможной электронной плотностью. Поэтому направление замещения водорода в бензольном кольце сильно зависит от природы уже имеющихся заместителей, которых делят на три группы, соответственно их влиянию на ход реакции замещения:
83
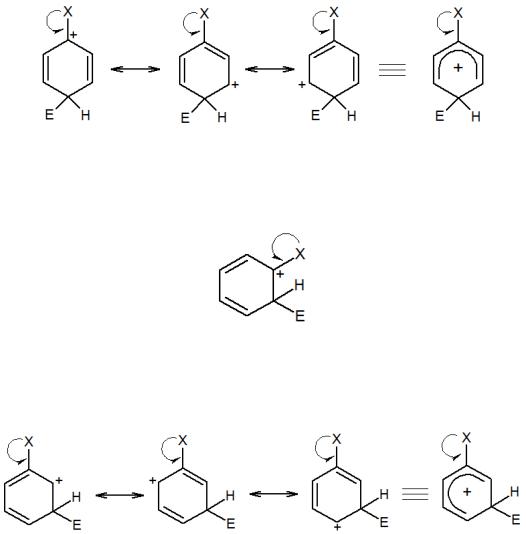
1. Ориентанты 1-го рода, 1-ой группы: орто-пара-ориентанты, активирующие ядро. Мезомерный эффект (+M) в этих группах сильнее, чем индуктивный эффект (-I) (в случае алкильных заместителей – это положительный индуктивный эффект и сверхсопряжение). Такие заместители активируют все положения в бензольном кольце, но особенно ускоряют замещение в орто- и пара-положениях. Общее ускорение реакции связано со значительным электронодонорным эффектом, стабилизирующим промежуточные σ-комплексы, особенно орто- и пара-σ- комплексы:
В этом случае первая структура является преобладающей ввиду непосредственной близости заместителя, компенсирующего положительный заряд. Для орто-σ- комплекса имеется подобная структура:
Одновременно с этим скорость замещения в мета-положение будет заметно ниже, поскольку группа Х и положительный заряд в резонансных структурах удалены друг от друга:
2. Ориентанты 1-го рода, 2-ой группы: орто-пара-ориентанты, дезактивирующие ядро, с сильным индуктивным эффектом ((+M) < ( -I)), понижающим электронную плотность в кольце, с чем связано снижение общей скорости замещения. При этом мета-положение дезактивируется сильнее всего, поскольку в орто- и пара-σ-комплексах присутствует положительный эффект сопряжения, которого нет у мета-комплекса:
84
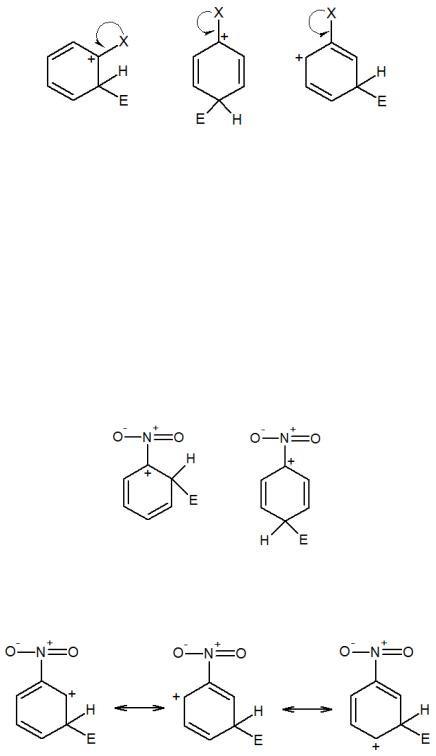
3. Ориентанты 2-го рода: мета-ориентанты, дезактивирующие ядро. Это атомные группировки, способные оттягивать (принимать) электроны от бензольного ядра, обладающие электроноакцепторными свойствами ввиду наличия целого или частичного положительного заряда на атоме, непосредственно связанном с углеродным атомом ароматического ядра. Преимущественное замедление скорости реакции замещения в данном случае будет наблюдаться в орто- и пара-положениях, что связано с сильной дестабилизацией положительного заряда в таких σ-комплексах. Например, в некоторых резонансных структурах орто- и пара-нитрозамещенных σ-комплексов два положительных заряда оказываются на соседних атомах:
мета-замещенные σ-комплексы дестабилизированы меньше, так как положительные заряды находятся на бо́льшем расстоянию друг к другу:
Примеры различных заместителей даны ниже в таблице. В действительности же необходимо понимать, что резкая граница между этими тремя группами отсутствует, и для каждого замещенного бензола образуются в различных соотношениях все возможные изомеры.
85

Примеры различных групп заместителей в ароматическом кольце
Заместители
1-го рода 1-ой группы |
1-го рода 2-ой группы |
2-го рода |
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
CH |
3 |
; C H |
; C |
H |
; -OH; -O-; |
-Cl; -F; -Br; -СН |
Сl; |
-Cl; -F; -Br; -СН |
Сl; |
|
|
2 |
5 |
6 |
5 |
|
2 |
|
2 |
|
|
-OR; -OC6H5; -NR2 ; -NH2; |
-CH=CH2 (-CH=CHR); |
-CH=CH2 (-CH=CHR); |
||||||||
-NHCOCH3, (-NHCOR). |
-C CH. |
|
-C CH. |
|
||||||
|
|
|
|
|
|
|
|
|
|
|
Реакционная способность замещенных бензолов для электрофильных реакций описывается уравнением Гаммета, но вместо стандартных констант заместителей σ правильнее использовать так называемые электрофильные константы σ+, учитывающие влияние не только индуктивного, но и резонансного эффекта заместителя (см. главу 1):
lg (ki/k0) = ρ·σ+.
Константа ρ, характеризующая чувствительность реакции к электронным влияниям заместителей, всегда меньше нуля, что может служить подтверждением механизма электрофильного замещения, ускоряемого электронодонорными заместителями. В то же время величина ρ зависит от активности электрофила, атакующего ароматическое ядро. Чем более активным является электрофил, тем более безразлично ему, какую молекулу или какое положение в ней атаковать, и тем меньше влияние заместителей и абсолютное значение ρ.
3.3. Механизм электрофильного замещения в алифатическом ряду
Общий вид реакций электрофильного замещения в алифатическом ряду мало отличается от нуклеофильного замещения. Однако существенная разница состоит в том, что вместо карбкатиона R+ должен образовываться карбанион R-, а замещаться должен положительно заряженный фрагмент молекулы. Карбанионы являются малоустойчивыми и реакционноспособными частицами, легко взаимодействуют с электрофилами и присоединяются по кратным связям. Очевидно, что стабильность их будет возрастать при наличии электроноакцепторных заместителей, а также сопряжённых кратных связей за счет делокализации заряда. Концентрация свободных карбанионов в растворе обычно крайне мала, и существуют они в виде контактных или сольватно-разделённых ионных пар. Поэтому очень редко
86
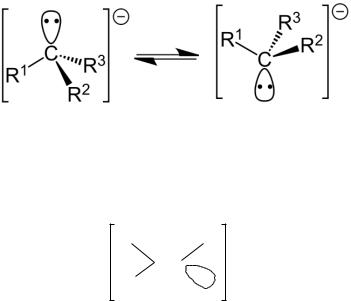
встречаются реакции, протекающие по мономолекулярному механизму с образованием карбанионов как кинетически независимых частиц.
Наиболее распространенными реагентами в реакциях электрофильного замещения в алифатическом ряду являются так называемые С–Н-кислоты. Это соединения, в которых С–Н-связь может гетеролитически расщепляться с образованием протона и соответствующего карбаниона. Центральный атом углерода в карбанионах предельного ряда сохраняет sp3-гибридизацию. Отрицательный заряд сконцентрирован в свободной электронной паре карбаниона, которая, согласно постулату Гиллеспи, стремится занять свое место в пространстве, подобно обычным -связям. Именно это обстоятельство обусловливает равновесное существование стереоизомеров (инверсию):
Карбанионы винильного типа сохраняют sp2-гибридизацию и существуют в цис- и транс-конфигурации. При диссоциации С–Н-связи у ненасыщенного соединения сохраняется его первоначальная конфигурация.
R1 R2 -
R3 C=C :
Cила С–Н-кислот зависит от характера заместителей у атома углерода: чем больше количество и сила электроноакцепторных заместителей, тем сильнее кислота, и тем легче образуется карбанион. Об этом свидетельствуют значения pKa сопряженных карбаниону кислот:
Карбанион |
R3C:— |
R3C:— |
ArCH2:— |
HC≡C:— |
HC≡C:— |
HC≡C:— |
рКа |
|
|
|
|
|
|
сопряженной |
40 |
36 |
34 |
25 |
11 |
7 |
кислоты |
|
|
|
|
|
|
|
|
|
|
|
|
|
Конечно, реагентами в реакции электрофильного замещения в алифатическом ряду могут быть не только С–Н-кислоты, но и соединения, содержащие
87
молекулярный фрагмент, соответствующий карбаниону. Например, магнийорганические соединения:
CH3-MgBr :CH3- + MgBr+
Находящиеся в реакционной среде основания (В) способствуют образованию карбаниона:
R3CH + B- [R3C:- HB]
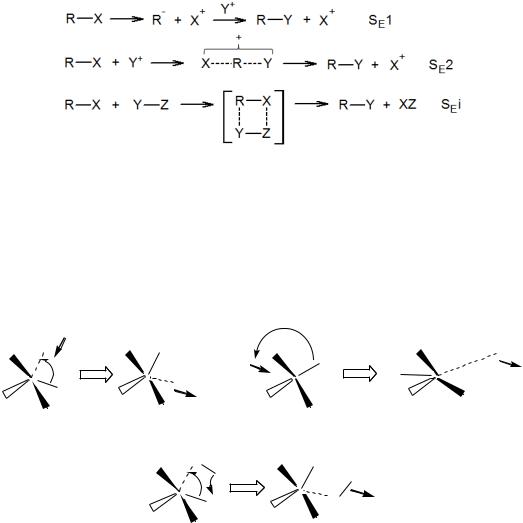
Возможны три механизма реакций электрофильного замещения при алифатическом углероде:
SE1 – мономолекулярное электрофильное замещение; SE2 – бимолекулярное электрофильное замещение;
SEi (internal substitution electrophilic) – «внутреннее замещение»:
Главное отличие от нуклеофильного механизма заключается в том, что атака электрофила может осуществляться как с фронта SE2(front), так и с тыла SE2(back), что в результате может привести к различному стереохимическому результату: как рацемизации, так и инверсии конфигурации.
Ниже приведены схемы, иллюстрирующие механизмы электрофильного замещения в алифатическом ряду:
|
Y |
|
Y |
Y+ |
|
|
|
X |
|
|
|
|
X |
|
|||
C |
|
C |
X |
C |
Y |
C |
||
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
SE2(front) |
|
|
|
|
SE2(back) |
|
|
|
|
|
|
Y Z |
|
Y |
|
|
|
|
|
|
C |
Z |
|
|
|
|
|
|
C |
X |
X |
|
|
|
|
|
|
|
|
|
|
|
SEi(internal, внутри)
88
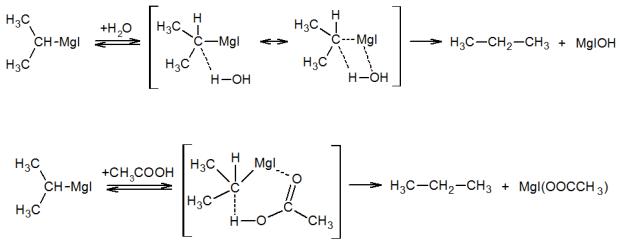
Для всех металлоорганических соединений (в том числе алкилмагнийгалогенидов, называемых реактивами Гриньяра) характерны реакции электрофильного замещения. Например, реакция изопропилмагний йодида с водой и уксусной кислотой могут протекать как по механизму SE2(front):
,
Так и по механизму SEi:
.
4. РАДИКАЛЬНЫЕ РЕАКЦИИ
Радикальные реакции — это процессы, идущие с участием радикалов, образующихся при гомолитическом разрыве ковалентной связи. Пара электронов, образующая связь, делится таким образом, что каждая из образующихся частиц получает по одному электрону. Разрыв связи и образование радикалов проходят намного легче в тех молекулах, в которых образующаяся частица может быть стабилизирована за счет эффектов сопряжения и гиперконъюгации (сопряжение с СН3 группой). Радикальные реакции включают две обязательные стадии — образование радикалов и их гибель. Если между этими двумя стадиями протекают циклические повторяющиеся процессы с участием образовавшихся радикалов – такая реакция называется цепной радикальной реакцией. Количество циклически повторяющихся процессов с участием образовавшихся радикалов называют длиной цепи. Если длина цепи равна 1, то говорят о радикальной не цепной реакции.
К типичной нецепной радикальной реакции можно отнести реакции цис- транс-изомеризации олефинов под действием УФ-света:
89

Квант света h переводит один электрон со связывающей орбитали олефина на разрыхляющую орбиталь *. В результате разрывается двойная связь и в образовавшемся бирадикале из-за электрон-электронного отталкивания происходит разворот левого или правого фрагмента на 90 градусов. Далее этот бирадикал стабилизируется при обратном переходе электрона с разрыхляющей орбитали * на связывающую орбиталь олефина с образованием вновь двойной связи.
|
|
|
|
|
|
h |
|
|
|
|
|
|
|
|
|
h |
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
цис - |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
транс - |
|||||||||||||
|
бирадикал |
|
|
|
|
|
||||||||||||||||||||||||
конфигурация |
|
|
|
|
|
|||||||||||||||||||||||||
конфигурация |
||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
Реакция цис-транс-изомеризации 2-бутена, предположительно, имеет следующий механизм:
CH |
CH |
|
CH |
CH |
CH3 |
CH3 |
3 |
3 |
|
3 |
|
|
|
|
|
|
|
3 |
|
|
|
* |
* |
* |
* |
|
|
|
CH3 |
|
CH3 |
|
|
|
|
|
|
|
|
|
Равновесие реакции при низких температурах смещено в сторону транс-изомера. Большинство радикальных реакций являются цепными и включают три стадии:
1)зарождение цепи – образование свободных радикалов;
2)продолжение цепи, в результате которого образуются продукты реакции и регенерируется исходный радикал:
90