

МЕХАНИЗМЫ МЕТОДИЧКА
.pdfВ теории абсолютных скоростей реакций Эйринг и Поляни приняли, что активированный комплекс находится в обычном термодинамическом равновесии с исходными реагентами. Это равновесие можно охарактеризовать активационными параметрами: свободной энергией активации G≠, энтропией активации S≠ и
энтальпией активации |
H≠, между которыми имеется обычная |
термодинамическая связь: |
|
|
G≠ = H≠ − T · S≠. |
Совместное решение уравнений равновесия активированного комплекса и, согласно статистической термодинамике, скорости его распада в продукты реакции
( G≠ = -RTln∆K≠)
τ h
кT ,
(τ – время распада активированного комплекса, h – постоянная Планка, к – постоянная Больцмана) приводит к такому выражению для константы скорости элементарной реакции (уравнение Эйринга–Поляни):
21

|
кT |
|
ΔS |
|
|
ΔH |
|
||
k |
|
exp |
|
|
exp |
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
. |
|
h |
|
R |
|
|
R T |
|||
−
Из его сравнения с обычной формой уравнения Аррениуса (K = A ) видно, что предэкспоненциальный множитель последнего является функцией энтропии активации и температуры.
Энтропию активации можно найти из экспериментальных данных по кинетике, имея в виду, что ek/h = 5,662 ×10−10 с−1, а предэкспоненциальный множитель найден для концентраций, выраженных в моль/л.
Энтропия активации дает некоторое представление о механизме элементарных реакций, так как она связана с изменением упорядоченности системы при образовании активированного комплекса. Для бимолекулярных реакций эта упорядоченность возрастает, а энтропия активации имеет отрицательное значение. Наоборот, для мономолекулярных реакций переходное состояние из-за удлинения рвущихся связей становится менее упорядоченным, и энтропия активации приобретает положительное значение.
1.6. Сольватация, идеи Борна и Кирквуда
Сольватация – взаимодействие молекул растворенного вещества с молекулами растворителя, которое состоит в том, что молекула растворенного вещества оказывается окруженной сольватной оболочкой, состоящей из молекул растворителя, и:
-приводит к изменению свойств молекул в растворе;
-влияет на все физические и физико-химические процессы;
-определяет скорость реакций в растворах, положение равновесия и их механизм. Наиболее интенсивна сольватация ионов в растворах электролитов.
22
Сольваты – молекулярные образования постоянного или переменного состава, время жизни которых определяется характером и интенсивностью межмолекулярных взаимодействий.
Важнейшие термодинамические характеристики сольватации:
-энтальпия сольватации ∆Hc определяет тепловой эффект внедрения молекулы растворенного вещества в растворитель;
-энергия Гиббса сольватации ∆Gc определяет растворимость вещества:
∆Gc = ∆Hc – T∆Sc,
где ∆Sc – энтропия сольватации; Т – абсолютная температура; ∆Hc – энтальпия сольватации; ∆Gc – энергия Гиббса сольватации.
Структура ближайшего окружения частицы растворенного вещества характеризуется координационными числами сольватации, т.е. количеством молекул растворителя, участвующих вместе с частицей в диффузионном движении. Число сольватации зависит:
-от природы растворенной частицы и растворителя;
-используемого метода определения.
При исследовании влияния растворителя на кинетику реакций было установлено, что химическую реакцию нельзя рассматривать отдельно от среды. Теоретический расчет влияния растворителя на реакционную способность растворенного вещества сопряжен с большими математическими трудностями.
Впрочем, разработаны простые, в том числе модельные, подходы к расчету ∆Hc и ∆Gc, в частности макроскопический (континуальный) и микроскопический (дискретный) методы описания эффектов сольватации. Континуальные методы основаны на моделях Макса Борна и Джона Кирквуда.
Свободная сольватация молекулы в среде равна:
|
|
|
1 |
∞ (n+1)(1−ε) |
N |
N |
|
|
|
(rjrk)n |
|
|
||
∆G |
c |
= |
|
∑ |
|
∑ ∑ |
k=1 |
Q |
Q |
k |
|
P cosθ |
jk |
, |
2 |
|
|
||||||||||||
|
|
n=0 n+(n+1)ε |
j=1 |
j |
|
a2n+1 n |
|
|||||||
где а – радиус полости, вырезаемой в результате внедрения молекулы растворенного вещества в растворитель; , – эффективные заряды на j-м и k-м атомах этой молекулы, N – число атомов в ней; - полиномы Лежандра, описывающие соответствующие монопольные, дипольные и т.д. взаимодействия и эффекты более высоких порядков; - углы, образованные векторами rj и rk, определяющими положения атомов j и k.
23

Частными случаями данного уравнения являются уравнения для свободной
энергии сольватации иона ∆ 0 – уравнение Борна: |
|
|
|||||
∆G0 |
= – |
1 Q2 |
(1– |
1 |
), |
||
2 |
|
a |
ε |
||||
|
|
|
|
|
|||
где Q – заряд иона, ɛ – диэлектрическая постоянная.
Поскольку изменение свободной энергии сводится к изменению энергии активации реакции, то отношение константы скорости реакции в среде с разными диэлектрическими свойствами ki к константе скорости, выбранной для сравнения k0, в логарифмической форме будет иметь вид:
k |
i |
|
|
|
|
|
ln |
k |
const 1 |
ε |
. |
||
|
|
0 |
|
Существует также идея Кирквуда для взаимодействующих диполей. Диполь – это наличие в молекуле двух равных по абсолютной величине разноименных точечных зарядов (+е; -е), находящихся на некотором расстоянии друг от друга. Идея Кирквуда заключается в использовании уравнения поляризуемости (Р, уравнение Клаузиуса–Мосотти) для оценки влияния полярности среды на реакцию дипольных молекул:
P M d ε 1  ε 2 ,
ε 2 ,
где М – молекулярная масса, d – плотность.
Это уравнение является линейной функцией энергии взаимодействия молекул с растворителем: отсюда, используя соотношение Максвелла ɛ = n2, получаем:
k |
i |
|
|
|
|
n2 |
1 |
2n2 |
|
ε 1 |
|
ln |
|
|
|
const |
|
|
|
const |
|
||
|
|
k |
0 |
|
|
|
|
1 |
|
2ε 1 , |
|
|
|
|
|
|
|
|
|
где n – показатель преломления света.
Идея применима везде, где используются диполи.
Таким образом, явление сольватации заключается в том, что растворитель изменяет скорость химических реакций (до 109 раз), определяет относительную устойчивость таутомеров, конформеров, изомеров, влияет на механизм реакций. Положения кислотно-основных равновесий в значительной степени определяются сольватирующей способностью растворителя.
24

1.7. Принцип линейности свободных энергий. Корреляционные уравнения. Уравнения Гаммета и Тафта, изокинетические соотношения и их физический смысл
В целом, рассмотренные уравнения изменения констант скоростей реакций под влиянием растворителя характеризуют изменение свободных энергий активации при взаимодействии молекул под влиянием электростатических полей. Прежде всего, речь идет о свободной энергии активации G≠, об энтропии активации S≠ и энтальпии активации H≠, которые связаны между собою в уравнении
Гиббса:
G≠ = H≠ − T · S≠.
Прологарифмировав уравнение Эйринга и Поляни для зависимости константы
скорости от энергии Гиббса, получим: |
|
|
|
|
|
|||||
ln k |
|
kT |
|
|
|
|
|
|
|
|
i |
h |
ΔSi |
R |
ΔHi |
RT |
kT |
|
ΔGi |
||
|
|
|
|
h |
|
RT |
||||
Разница между уравнениями для одной реакции с константой скорости ki и какой-либо другой однотипной реакции k0 будет соответствовать разнице в свободных энергиях активации этих реакций:
|
|
k |
|
|
ΔG |
|
|
|
ΔG |
|
|
δΔG |
|
|
ln |
|
i |
|
|
0 |
|
|
i |
|
|||
|
|
|
|
|
k0 |
|
|
RT |
|
|
|
RT |
i,o . |
|
|
|
|
|
|
|
|
|
|
||||
Величина |
δΔG |
|
|
соответствует вкладу в энергию Гиббса, который внес |
|||||||||
|
i,o |
|
|
||||||||||
изменяемый параметр в этих реакциях, например, диэлектрические свойства растворителя. Основываясь на постулате, что энергия – аддитивная величина,
говорят о выполнении общего соотношения линейности свободных энергий
(ЛСЭ).
Для различных реакций, в которых меняется тот же параметр (например, в реакции этерификации кислот kj и диссоциации этих же кислот Kj в разных растворителях), изменение полярности растворителя приводит к одинаковому эффекту. Математически, этот эффект выражается в выполнении соотношения ЛСЭ:
25

ln k j α ln K j c ,
где с – постоянная.
На основе принципа линейности свободных энергий (ПЛСЭ) существует большое количество корреляционных зависимостей. Корреляционные уравнения устанавливают аналогии в воздействии выбранных факторов на химические или физические свойства системы.
Любую корреляцию, учитывающую влияние заместителя, растворителя и т.п. на химическую реакцию, можно представить в виде уравнения:
ln k j |
n |
ρ F параметр c . |
|
|
1 |
Левая часть уравнения содержит логарифм относительной константы скорости kj. Правая часть содержит, как правило, алгебраическую сумму членов, часть из которых, являются функциями эмпирических параметров ΣF(параметр), которые выражают изменение свободной энергии под воздействием заместителей в молекуле или растворителя. Коэффициент чувствительности реакции ρ зависит от типа реакции и температуры. Свободный член с численно соответствует логарифму константы скорости ln(kj) при отсутствии воздействия на реакцию изучаемых параметров. Каждый параметр вносит свой аддитивный вклад в свободную энергию, а соотношение этих вкладов остаётся пропорциональным для других процессов тех же реагентов.
Наиболее востребованными в органической химии корреляционными уравнениями являются уравнение Гаммета и уравнение Тафта.
Первое из них применимо к ароматическим соединениям. Оно устанавливает линейную связь между константами равновесий замещенных бензойных кислот и константами скоростей химических реакций ароматических соединений с теми же заместителями, т.е. между свободными энергиями этих процессов ( G0 или G≠ ). Таким образом, отражает принцип линейности свободных энергий (принцип ЛСЭ, установленный исключительно на основе экспериментальных данных). Уравнение Гаммета применимо не только к равновесным процессам, но и для необратимых реакций.
k |
i |
|
|
ρ σ |
|
lg |
k |
|
|
||
|
|
0 |
|
, |
26

где ki – константа скорости реакции соединения с заместителем; k0 – константа скорости реакции незамещенного соединения; ρ – чувствительность влияния заместителя; σ – константа заместителя.
Константа заместителя σ является термодинамической характеристикой реакции диссоциации замещенной бензойной кислоты и равна логарифму соотношения констант диссоциации бензойных кислот с заместителем Ki и без него K0 при 25 °С:
K |
i |
|
σ |
|
lg |
|
. |
||
|
|
K0 |
|
|
|
|
|
Уравнение Гаммета позволяет с точностью ± 15 % рассчитать кинетические параметры реакций производных бензола с пара- и мета-заместителями (σп и σм, соответственно). Для реакций, ускоряемых электронодонорными заместителями, σ <0 и ρ<0. Для реакций, ускоряемых электроноакцепторными заместителями σ >0
и ρ>0.
k |
i |
|
ρ σ |
|
lg |
|
. |
||
|
|
k0 |
|
|
|
|
|
Значения констант (σп и σм), которые были рассчитаны по данным констант диссоциации замещенных бензойных кислот, содержат информацию только об индуктивном эффекте. Поэтому, они могут оказаться значительно завышенными или заниженными, если реакционный центр ароматического соединения может взаимодействовать с заместителем по другим механизмам. Например, проявляется π-сопряжение между нитрогруппой в кольце и реакционным центром с ОНгруппой.
O
N
 OH
OH
O
Например, ОН-группа в орто-нитробензоле легко расщепляется с образованием таутомера и практически не проявляет свойств гидроксильной группы.
27

В связи с возможностью дополнительного воздействия на реакционный центр (резонансный эффект), появились дополнительные константы заместителей, как σ- и σ+. Их называют нуклеофильными и электрофильными константами, или «усиленными» донорами и акцепторами. Для учета резонансного эффекта π-связей используют σR-константы. Константа σI учитывает только индукционное влияние заместителя.
Уравнение Тафта применимо только для алифатических и алициклических соединений, которые не являются производными бензола. В этом случае сравнивают константы скоростей с константами кислотности замещенной уксусной кислоты при 25 °С.
k |
i |
|
ρ * σ * |
|
lg |
|
, |
||
|
|
kCH3 |
|
|
|
|
|
где σ* – константа Тафта; ρ* – параметр Тафта.
Константа Тафта σ* отражает индукционную составляющую влияния заместителя на константы скорости и линейно связана с «индуктивной» константой Гаммета σI соотношением:
σ* R 6,23 σI R ;
σI R 0,45 σ * CH2R .
Как уже понятно из строения объемных заместителей, их индуктивное влияние сравнимо или меньше, чем стерическое взаимодействие с реакционным центром, ввиду слишком малого расстояния между ними. Для учета пространственных эффектов заместителя ввели стерические константы Еs. Обычно используют двухпараметрическое уравнение Тафта, которое учитывает совместно влияние электронных и стерических факторов.
k |
i |
|
ρ * σ * ρ |
|
E |
|
lg |
|
s |
s . |
|||
|
|
kCH3 |
|
|
||
|
|
|
|
|
||
|
|
28 |
|
|
|
|

Более редко используют четырехпараметрическое уравнение Тафта, в которое добавлено влияние гиперсопряжения и резонансное влияние заместителя:
k |
|
|
|
|
lg |
i |
ρ * σ * ρsEs h |
Ψ |
|
|
|
kCH3 |
|
, |
где введены константы: h – гиперконъюгационное влияние; Δn – количество СН3 групп у реакционного центра; Ψ – резонансное влияние.
Изокинетические соотношения и их физический смысл. При рассмотрении кинетических закономерностей однотипных серий реакций (серия i реакций, например, реакции этерификации метанолом замещенных бензойных кислот) очень часто обнаруживают линейные корреляции между энергиями активации Ei,a и
предэкспоненциальными множителями Ai в уравнениях Аррениуса:
lg Ai α β Ei,a .
для реакций, где константа скорости реакции с i-заместителем выражается как
|
E |
|
|
|
|
|
|
|
|
i,a |
RT |
k(i) Ai e |
|
|
|
|
|
. |
Большинство исследователей предпочитают работать с несколько иной, более информативной корреляцией между энтропиями и энтальпиями активации однотипных серий реакций:
ΔSi α β ΔHi
Если сравнивать оба выражения с точки зрения теории абсолютных скоростей, то видна их идентичность. Для ряда однотипных реакций рассмотренные параметры процессов – энтальпии и энтропии активации – строго закоррелированы. Этот многократно обсуждаемый эмпирический факт получил название
«компенсационный эффект», чаще подчеркивают компенсационный эффект энтальпия - энтропия или говорят об изокинетическом соотношении энтальпии
– энтропии. Относительно природы компенсационного эффекта до сих пор не утихают споры.
Для выяснения физического смысла подобных корреляций воспользуемся теоремой:
29
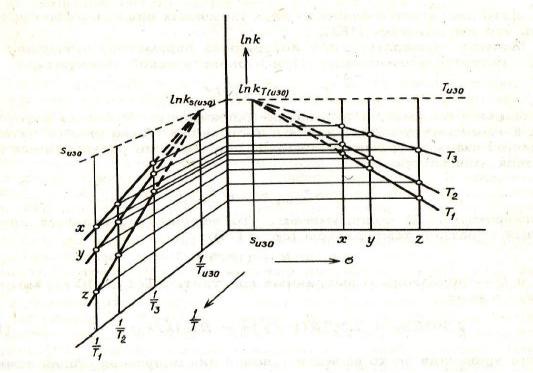
Если выполняется линейность корреляционной зависимости логарифма констант скоростей и какого-либо параметра реакции (из уравнений Гаммета, Тафта и т.д.) при двух температурах, то точка пересечения этих зависимостей будет общей точкой пересечения для этих же корреляций при других температурах. Теорема справедлива для диапазона температур, в котором выполняется закон Аррениуса.
Рис. 1.5 Соотношение между графиком Аррениуса и корреляцией Гаммета
Координатами точки пересечения корреляций будут {Ln(kТизо), изо}, которые соответствуют «изокинетической» константе скорости и «изокинетическому» параметру изо. Ордината точки пересечения соответствует логарифму константы скорости реакции, характеризуемой параметром изо для любой температуры, или для любого другого параметра, но при так называемой изокинетической температуре Тизо (рис. 1.5).
То же самое можно получить и для аррениусовского графика, поскольку реакции одной серии дают набор прямых, пересекающихся в одной точке с абсциссой, равной Тизо. Совпадение численных значений Тизо, полученных по двум графикам, убедительно доказывает наличие эффекта, который называют изокинетическим соотношением (ИКС) или эффектом компенсации. Величину Тизо (размерность градусы Кельвина) называют температурой компенсации.
30