

МЕХАНИЗМЫ МЕТОДИЧКА
.pdf3.1.1. Механизм электрофильного присоединения к олефинам
Первым этапом электрофильного присоединения всегда остается электрофильная атака двойной связи. Поэтому для большинства реакций типичен кислотный катализ. Он осуществляется двумя основными путями: первичным присоединением протона к двойной связи с первоначальным образованием -комплекса или активированием реагента с образованием более сильного электрофила, который способен присоединиться к двойной связи. В обоих случаях получается карбкатион, который, далее, взаимодействует с нуклеофилом.
Общая для всех процессов реакция электрофильного присоединения протона или другого электрофила по двойной связи не является одностадийной. Присоединению всегда предшествует образование -комплекса электрофила с олефином. При образовании -комплекса происходит орбитальное взаимодействие свободной - орбитали электрофила (Е+) с -связывающей орбиталью олефина:
|
E+ |
|
|
|
|
|
|
|
|
|
|
E |
||
|
||
|
E+ |
|
E+ |
|
|
|
|
|
Орбиталь образующегося -комплекса |
( ( , )) является связывающей и |
нахождение на ней электронной плотности, перенесенной с -орбитали олефина приносит выигрыш в энергии, т.е. энергию стабилизации -комплекса Е. Эта энергия не высока, составляет 4–21 кДж/моль, что не дает комплексу быть устойчивым. Они легко распадаются на исходные компоненты.
Акцепторными свойствами обладают также свободные низколежащие орбитали других электрофилов: например, иона NO2+, катион-радикала R+, поляризованных молекул галогенов Hal + Hal -, включая их в составе молекул кислот Льюиса:
71
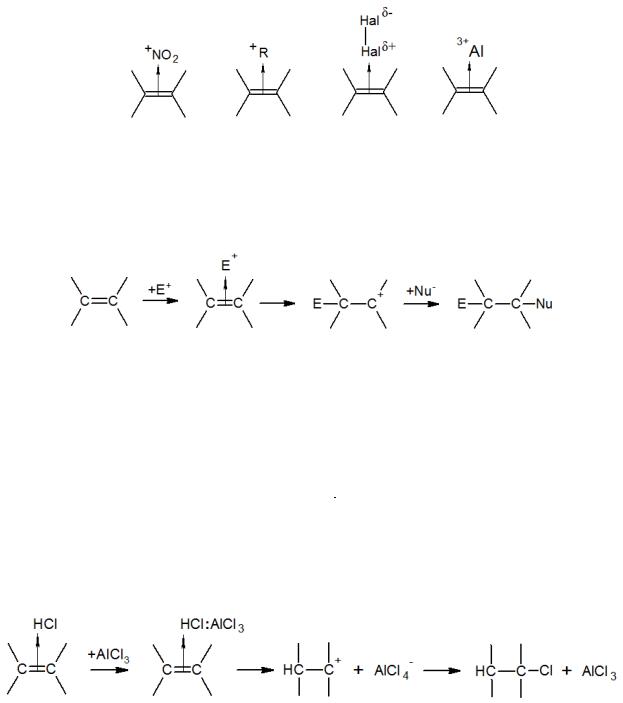
Дальнейшее превращение π-комплекса в карбкатион происходит по медленной мономолекулярной реакции, в результате чего электроны π-связи полностью переходят на свободные орбитали одного из атомов углерода и электрофила и образуется σ-связь С–Е или С–Н:
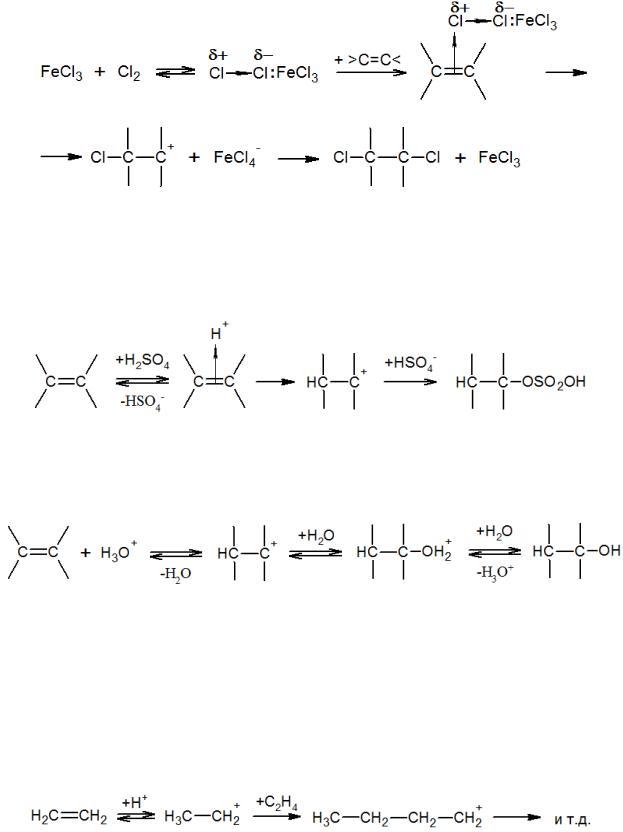
Некоторые реакции электрофильного присоединения, такие как гидрогалогенирование и галогенирование, ускоряются кислотами Льюиса (FeCl3, SnCl4, TiCl4 и т.д.). Механизм катализа гидрогалогенирования заключается в увеличении кислотности галогеноводорода в результате связывания иона галогена в комплекс с кислотой Льюиса, например:
HCl + SnCl4 
 H+SnCl5-
H+SnCl5-
Сопряженная кислота является более мощным протонодонором, чем галогеноводороды. Кислоты Льюиса также могут воздействовать на уже образовавшийся π-комплекс, ускоряя его переход в ион карбония:
При галогенировании происходит поляризация галогена, которая усиливается при взаимодействии галогена с кислотами Льюиса:
72
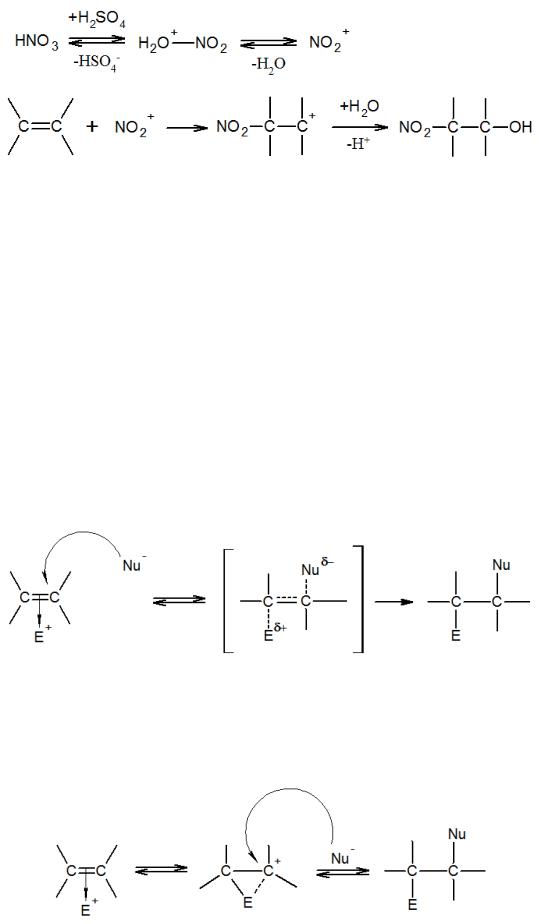
Присоединение сильных кислот проводят без добавления дополнительных каталитических веществ, так как в данном случае сама кислота выступает в роли катализатора и нуклеофильного реагента. В качестве примера можно привести присоединение серной кислоты:
Напротив, присоединение слабых электрофилов протекает только в присутствии каталитических добавок сильных протонных кислот. Так, для присоединения воды к олефинам в качестве катализатора используют серную или фосфорную кислоту:
По такому же механизму к олефинам присоединяются сероводород, меркаптаны, спирты, фенолы, карбоновые кислоты и пр. Скорость всех этих реакций определяется скоростью образования карбкатиона и прямо пропорциональна кислотности среды.
Кислотная полимеризация олефинов также является реакцией электрофильного присоединения. Роль нуклеофила, атакующего карбкатион, в этом случае выполняет сам олефин:
При нитровании олефинов смесями серной и азотной кислот электрофильным агентом является образующийся катион нитрония:
73
Фактически, превращение -комплекса может идти по двум путям, в зависимости от природы реагентов и условий протекания процесса:
-либо он превращается в карбкатион по медленной мономолекулярной реакции и далее быстро реагирует с нуклеофилом, регенерируя катализатор;
-либо по медленной бимолекулярной реакции поляризованный фрагмент олефина атакуется нуклеофилом, регенерируя катализатор.
По этой причине стереохимия реакций электрофильного присоединения различна. Необходимо особенно отметить довольно частое образование транс- продуктов, что, как правило, объясняют протеканием реакции по механизму, в котором лимитирующей стадией является бимолекулярное взаимодействие π- комплекса с нуклеофилом. Как следствие, нуклеофильная частица на второй стадии процесса может осуществлять атаку только с противоположной стороны от электрофила:
Стереоспецифичность реакции транс-присоединения, особенно часто встречаемую при галогенировании, объясняют также образованием «неклассических» ионов карбония, в которых сохраняется жесткая трехцентровая связь, препятствующая свободному вращению вокруг С–С-связи:
74
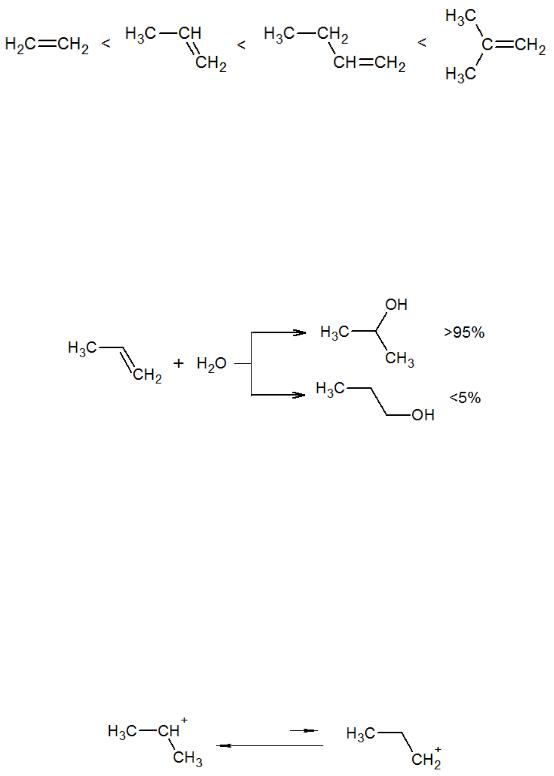
Как уже было показано в предыдущей главе, электронодонорные заместители в субстрате стабилизируют карбкатион и тем самым ускоряют реакцию присоединения. Таким образом, реакционная способность к электрофильному присоединению в ряду алкенов с электронодонорными заместителями меняется в ряду:
Так, например, гидратация изобутилена проходит в 8000 раз быстрее, чем гидратация пропилена, который реагирует в 8000 раз быстрее этилена.
Электронными эффектами, влияющими на стабилизацию карбкатиона, определяется правило электрофильного присоединения к несимметричным олефинам. Например, при гидроксилировании пропилена преимущественно образуется только один изомер – изопропиловый спирт. Доля н-пропанола весьма незначительна.
Такую избирательность заметил Марковников, сформулировав правило: «при присоединении протонных кислот и воды к несимметричным алкенам и алкинам атом водорода присоединяется к наиболее гидрогенизированному атому углерода».
Объяснение этого правила достаточно простое: реакция протекает через стадию образования карбкатиона. Поэтому, чем стабильнее ион, тем быстрее идет реакция. Если есть возможность образования двух ионов, то их соотношение будет зависеть от стабильности каждого иона. Чем более стабилен один из них, тем выше его концентрация и тем большее количество продуктов получается с его конфигурацией. При гидратации пропилена образуется, в основном, более стабильный вторичный карбкатион:
75
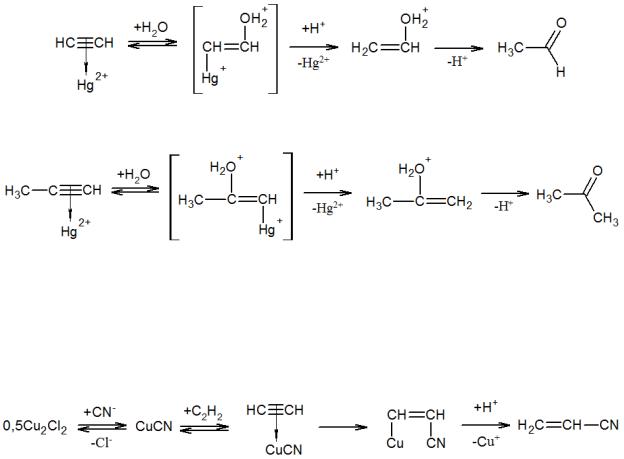
3.1.2. Реакции присоединения к ацетилену
Вотличие от олефинов, алкины, имеющие тройную связь, труднее вступают в реакции электрофильного присоединения. Это связано с тем, что тройная связь является более короткой, и π-электроны удерживаются на ней сильней, чем на двойной связи алкенов, что в конечном итоге приводит к снижению поляризуемости связи.
Реакции присоединения к алкинам катализируются солями и комплексами переходных металлов, особенно ртути (HgSO4, HgCl2), одновалентной меди (Cu2Cl2), цинка, кадмия, никеля, палладия. Например, в реакции Кучерова молекула ацетилена вначале активируется катионом ртути, после чего углеродный атом атакуется молекулой воды с последующим протолизом связи С–Н и изомеризацией
вацетальдегид. Реакция проводится в кислой среде, что обеспечивает немедленный протолиз металлоорганического переходного состояния с регенерацией катализатора:
Вслучае гидратации гомологов ацетилена образуются кетоны:
Аналогично протекают реакции при катализе солями кадмия и цинка. Хлорид меди используют для гидроцианирования ацетилена. Этот процесс был одним из первых промышленных методов синтеза акрилонитрила. Механизм реакции сходен с рассмотренным выше, отличие заключается лишь в атаке углеродного атома цианид-анионом:
Наличие кратных π-связей между атомами углерода обусловливает возможность двухступенчатого последовательного присоединения уже двух молекул X-Y. Так
76
же, как и в реакции присоединения к олефинам, в результате образуются транс- изомеры.
Как и олефины, ацетиленовые соединения также могут вступать в реакции олигомеризации (полимеризации), в частности, известен механизм димеризации ацетилена. Реакция протекает при катализе солью меди через последовательное образование ацетиленида из π-комплекса, который затем образует второй π- комплекс с другой молекулой ацетилена. Дальнейшее взаимодействие протекает внутримолекулярно по так называемому механизму внедрения по металлуглеродной связи:
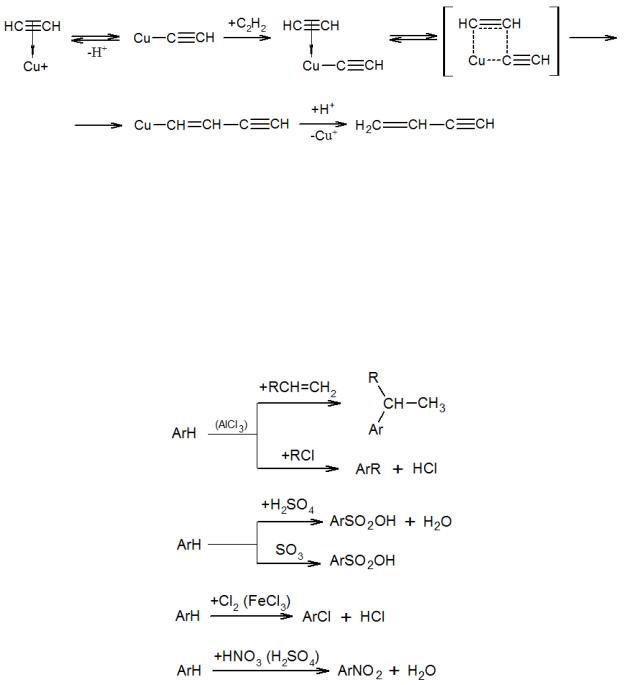
3.2. Электрофильное замещение в ароматических соединениях
(SEAr)
Среди многих реакций электрофильного замещения в ароматических соединениях в основном органическом и нефтехимическом синтезе главное значение имеют реакции алкилирования, хлорирования, нитрования и сульфирования:
77
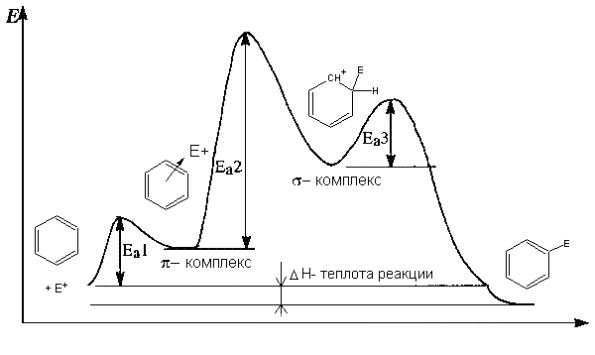
Механизм реакций идентичен механизму электрофильного присоединения по кратным связям. Реакция начинается с образования неустойчивого -комплекса электрофила Е+ с ароматическим ядром. Реакция имеет малую энергию активации Еа1. В комплексе электрофил не локализован на каком-то атоме углерода, а связан со всеми -электронами ароматического кольца. Далее этот -комплекс превращается в -комплекс. Это превращение требует больших затрат энергии Еа2
(рис. 3.7).
Рис. 3.7 Схема электрофильного замещения в ароматических соединениях SEAr
Образовавшийся -комплекс перегруппировывается в продукты реакции с небольшой энергией активации Еа3.
3.2.1. Реакции галогенирования
Рассмотрим более детально механизм реакции галогенирования, например, бромирования бензола в присутствии катализатора АlBr3. В реакционной среде катализатор, координируя молекулу брома, образует электрофильный атом брома:
Br2 + AlBr3 Br + [AlBr4] -
Первой стадией электрофильного замещения является образование π-комплекса в результате взаимодействия π-электронной системы бензольного кольца с
78
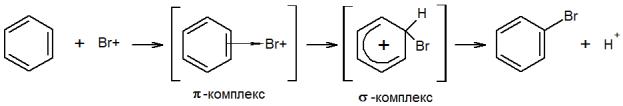
положительно-заряженным электрофилом - с Br+ При этом атакующая частица связывается со всеми шестью π-электронами кольца.
Вторая стадия состоит в переходе π-комплекса в σ-комплекс. Это происходит в результате выделения из системы шести π-электронов двух электронов для образования новой ковалентной связи С—Br. Оставшиеся четыре π-электрона распределяются между пятью углеродными атомами бензольного кольца:
Разорванная окружность в цикле означает прерванное π-сопряжение электронов. σ-Комплекс – промежуточное образование, представляющее собой
неустойчивый карбкатион (но более устойчивый, чем π-комплекс), лишенный ароматичности. Шесть его углеродных атомов находятся в различных валентных состояниях: один, с которым связан заместитель Br, – насыщенный (в состоянии sр3-гибридизации), а пять других – в обычном для бензола втором валентном состоянии (sp2). Заместитель Br и атом водорода при насыщенном углеродном атоме расположены в плоскости, перпендикулярной плоскости бензольного кольца.
Третья стадия – быстрое отщепление протона от σ-комплекса. Освобождающаяся при этом пара электронов (от связи С–Н) восполняет электронный пробел в кольце, и углерод из состояния sp3 переходит в состояние sр2-гибридизации. Отщепившийся протон связывается с анионом Br-, обычно находящимся в комплексе катализатора, например (АlBr4-).
По такому же механизму протекают все реакции электрофильного замещения в бензольном ядре – галогенирование, нитрование, сульфирование, реакции алкилирования и ацилирования. В зависимости от условий одни и те же реагенты могут взаимодействовать и с ароматическим ядром, и с боковой цепью.
3.2.2. Реакция нитрования
Реакцию нитрования ароматических соединений проводят так называемой нитрующей смесью – смесью концентрированных азотной и серной кислот (при температурах, не превышающих 40–50 °С). При взаимодействии этих кислот образуется смесь ионов, из которых нитрующим агентом является ион нитрония
NO2+:
79
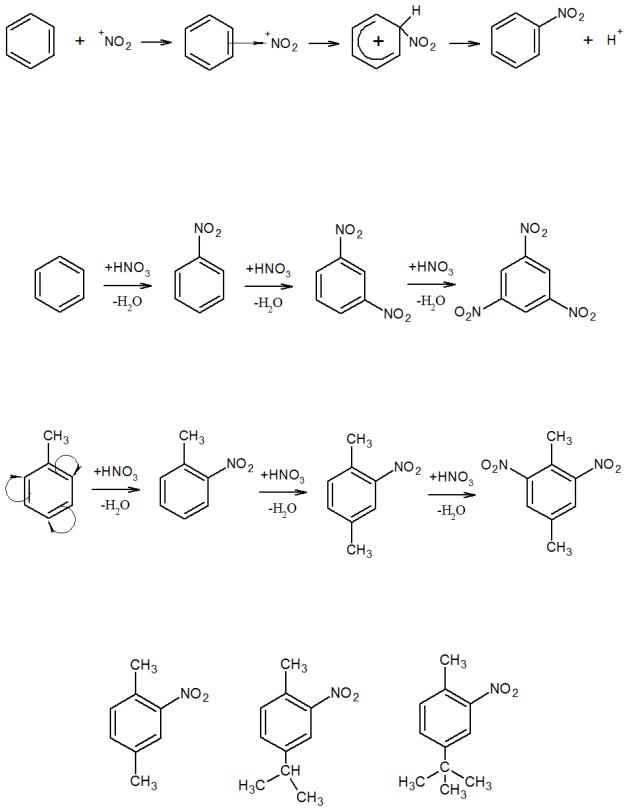
HNO3 + 2H2SO4 NO2+ + H3O+ + 2HSO4-
Ион нитрония атакует бензольное кольцо, образуя последовательно – и – комплексы и, наконец, продукты реакции:
При введении второй нитрогруппы в ароматическое ядро находящаяся в бензольном кольце нитрогруппа уменьшает скорость дальнейшего нитрования. Однако в жестких условиях можно ввести вторую (при 90 °С) и даже третью (при 100–110 °С) нитрогруппы, которые располагаются в мета-положениях относительно друг друга:
Толуол нитруется во много раз легче, чем бензол. Это объясняется +I - эффектом метильной группы (создается избыток электронной плотности в кольце в орто- и пара-положении):
При нитровании гомологов бензола, содержащих два заместителя, сказывается стерический эффект. Если, например, эти заместители находятся в пара-положении, нитрогруппа становится рядом с меньшим заместителем:
80