

МЕХАНИЗМЫ МЕТОДИЧКА
.pdfстроении на скорость реакции по-разному будут влиять и заместители при реакционном центре.
Очевидно, что протеканию реакции SN1 будут способствовать факторы, стабилизирующие промежуточный карбкатион, а именно наличие электронодонорных заместителей, их эффекты сопряжения и сверхсопряжения. Поэтому скорость будет возрастать при увеличении количества алкильных групп у реакционного центра. Склонность к замещению по SN1-механизму проявляют также соединения с сопряженными винильными и фенильными группами за счет образования единой π-сопряженной системы. Таким образом, реакционная способность к мономолекулярному нуклеофильному замещению растет в следующем ряду:
Следует отметить влияние кислородсодержащих заместителей. В α-положении к уходящей группе такой заместитель дает эффект сопряжения с реакционным центром, и SN1-реакция облегчается, в то время как при β-положении кислородсодержащей группы субстрат заметно менее реакционноспособен ввиду отсутствия сопряжения:
Внекоторых случаях образованию карбкатиона способствует образование внутримолекулярной водородной связи, вызванное особым расположением функциональных групп, как, например, в α-галогенкарбоновых кислотах:
ВSN2-реакциях центральный углеродный атом несет частичный отрицательный заряд и стабилизируется электроноакцепторными заместителями, способствующими рассредоточению этого заряда и уменьшению энергии активированного комплекса и, как следствие, ускоряют реакцию. Поэтому легко
41

предположить, что строение углеводородного радикала будет прямо противоположно влиять на протекание SN2-реакций, в отличие от мономолекулярного замещения. Уменьшение числа метильных групп при реакционном центре будет увеличивать скорость реакции, одновременно с этим снижая стерические трудности для атаки нуклеофилом. В результате получается, что первичные алкилзамещенные реагируют по SN2-механизму, а третичные – по SN1, а между крайними случаями находится область пограничных механизмов.
2.1.2. Влияние замещаемой группы
Нуклеофильное замещение как по SN1-, так и по SN2-механизму протекает тем быстрее, чем ниже энергия гетеролитического разрыва связи С–Х. Это во многом связано со стабилизацией образующегося аниона за счет его лучшей поляризуемости (в случае галоген-анионов) или эффектов сопряжения (для эфиров сульфокислот и диалкилсульфатов):
Кислые эфиры серной кислоты обладают лишь слабой алкилирующей способностью, так как у них легче разрывается связь S–O. Спирты и простые эфиры, как мы видели выше, реагируют только при кислотном катализе, когда поляризуется связь С–О:
2.1.3. Влияние нуклеофильного реагента
Скорость реакций нуклеофильного замещения определяется степенью нуклеофильности реагента, которая в свою очередь зависит от его основности и поляризуемости.
42
Влияние основности становится понятным из самой сути реакций нуклеофильного замещения: нуклеофил атакует субстрат, имеющий дефицит электронов; при этом сам нуклеофил должен обладать неподеленной электронной парой или отрицательным зарядом, то есть повышенной электронной плотностью. Очевидно, что чем выше электронная плотность на нуклеофильной частице, тем более охотно идет замещение. Влияние основности обычно наиболее сильно проявляется в реакциях, идущих по SN2-механизму, а также по пограничным механизмам, которые близки к SN2. При переходе к SN1-механизму наблюдается снижение влияния основности нуклеофила, поскольку образование новой связи происходит на все большем расстоянии. В конечном итоге, при «чистой» SN1- реакции скорость совсем не зависит от основности реагента. Количественно эта зависимость выражается уравнением Бренстеда, которое является частным случаем линейного соотношения свободных энергий:
k = GKbβ или lgk = lgG + βlgKb |
или lg |
ki |
= βlg |
Kb,i |
, |
|
|
||||
|
|
k0 |
|
Kb,0 |
|
где k – константа скорости, Kb - константа основности нуклеофила, G и β – эмпирические константы, k0 и Kb,0 – константы скорости и основности для стандартного вещества.
По зависимости относительной скорости реакции lg |
ki |
от относительной |
|||
k |
|||||
|
|
|
|
||
|
|
0 |
|
||
основности lg |
Kb,i |
можно судить о влиянии основности нуклеофила на скорость |
|||
Kb,0 |
|||||
|
|
|
|
||
реакции замещения и, соответственно, о механизме реакции. Чем выше β, тем больше тангенс угла наклона линейной зависимости, тем в большей степени проявляется зависимость скорости замещения от основности нуклеофила. На рис. 2.1 показан пример изменения относительной реакционной способности алкоголятов в реакции с некоторыми хлорпроизводными:
RCl + RiO- → RORi + Cl-
Степень зависимости скорости замещения от основности нуклеофила зависит от строения субстрата. Выше было показано, какие соединения проявляют склонность к реагированию по SN2- и SN1-механизмам. По графику также видно, что скорость замещения галогена в первичных хлорпроизводных, реагирующих преимущественно по бимолекулярному механизму, сильно зависит от относительной основности алкоголят-аниона. Появление в молекуле субстрата заместителей, проявляющих эффекты сопряжения и гиперконъюгации, постепенно
43
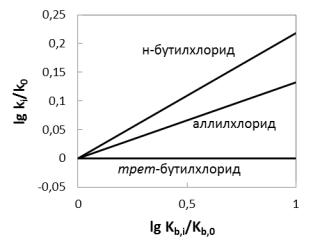
переводит механизм реакции на SN1, который, в предельном случае (напр., третичные галогенпроизводные), совсем не зависит от основности нуклеофила.
Рис. 2.1 Зависимость относительной реакционной способности алкоголятов от их основности для реакций с органическими хлоридами
Помимо основности на скорость нуклеофильного замещения влияет поляризуемость реакционного центра нуклеофила. В поляризованных молекулах электронная плотность может легко смещаться в сторону положительного заряда, образуя связь с молекулой субстрата на большом расстоянии и быстро реагируя с ней. Нуклеофилы, не способные поляризоваться, либо поляризуемые очень слабо, будут реагировать медленнее, чем сильно поляризованные ионы, даже если основность последних на порядки ниже. Например, средняя относительная нуклеофильность легко поляризуемых ионов C6H5S- в 470 раз выше, чем для ионов С2H5O-, тогда как основность иона С2H5O- (pKb = -2) на несколько порядков выше, чем C6H5S- (pKb = 4,6). Как правило, хорошей способностью к поляризации обладают ионы большого радиуса (S-, I-, Br-), а также частицы с кратными связями
(N3-, CN-).
Для количественной оценки нуклеофильности Эдвардс предложил следующее уравнение:
E = 3,6 lg |
Ri |
+ 0,0624 (pK |
a |
+ 1,74), |
|
||||
N |
RH O |
|
||
|
2 |
|
|
|
где R – рефракция (поляризуемость), а pKa – кислотность кислоты, сопряженной нуклеофилу. Стандартом служит вода, для которой нуклеофильность принята равной нулю.
Как правило, замещение с сильными нуклеофилами протекает по SN2-механизму, со слабыми же более быстро реакция проходит по пути SN1. Таким образом, наблюдается обращение механизмов, причем в области механизмов SN1 скорость не зависит от природы нуклеофила (рис. 2.2).
44
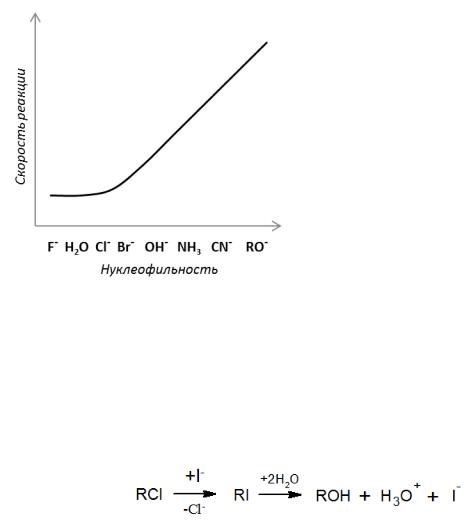
Рис. 2.2 Зависимость скорости реакции замещения от нуклеофильности реагентов
Установлено, что некоторые легко поляризуемые нуклеофилы одновременно являются легко уходящими группами, что позволяет использовать их в качестве нуклеофильных катализаторов. Например, анион йода сильно ускоряет относительно медленные реакции гидролиза алкилхлоридов водой. На первой стадии происходит замещение хлорид-аниона йодом, на второй – гидролиз образовавшегося алкилиодида с получением спирта и регенерацией йод-аниона:
2.1.4. Конкуренция нуклеофилов при замещении
Часто при проведении реакции замещения в реакционной массе находятся несколько нуклеофилов, обусловливающих параллельное образование нескольких продуктов. Кроме того, некоторые нуклеофилы могут иметь несколько реакционных центров. Преимущественное образование того или иного продукта происходит соответственно правилу Корнблюма: при SN2-замещении органический субстрат реагирует преимущественно с реагентом, имеющим бо́льшую нуклеофильность, зависящую от основности и поляризуемости; при SN1- замещении карбкатион обычно менее избирателен, но все же быстрее реагирует с нуклеофилом, имеющим наибольшую электронную плотность на реакционном центре (как правило, у наиболее электроотрицательного атома).
Для примера рассмотрим реакции с нуклеофилами, обладающими двойственной реакционной способностью: CN-, NO2- и др. Они обычно дают смесь продуктов замещения, но соотношение продуктов будет также зависеть от природы субстрата
45
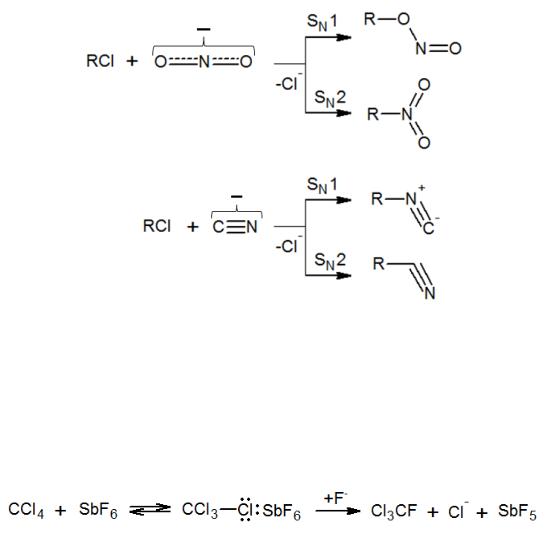
(в том числе замещаемой группы), во многом определяющей механизм реакции. В зависимости от этого будут преимущественно образовываться следующие продукты:
Похожий случай наблюдается при обмене галогенов. Реакция SN2 всегда протекает в сторону замещения более электроотрицательного атома на более нуклеофильный (F и Cl на Br и I). Однако при SN1-замещении образуется продукт с более электроотрицательным заместителем (т.е. RF из RCl). Для изменения механизма используют соли или катализаторы с сильно электрофильным катионом
(AgF, HgF2, SbF3, SbF5).
2.2. Нуклеофильное замещение при атоме углерода в ароматическом ядре
Нуклеофильное замещение в ароматическое ядро встречается крайне редко. Протекание таких реакций затруднено как по механизму SN1 (из-за малой стабильности фенильных катионов), так и по механизму SN2 (из-за повышенной электронной плотности на атоме углерода в ядре). Единственным примером замещения по механизму SN1 являются реакции с катионами диазония. Образование нестабильного фенил-катиона компенсируется энергетически выгодным образованием молекулы азота:
46
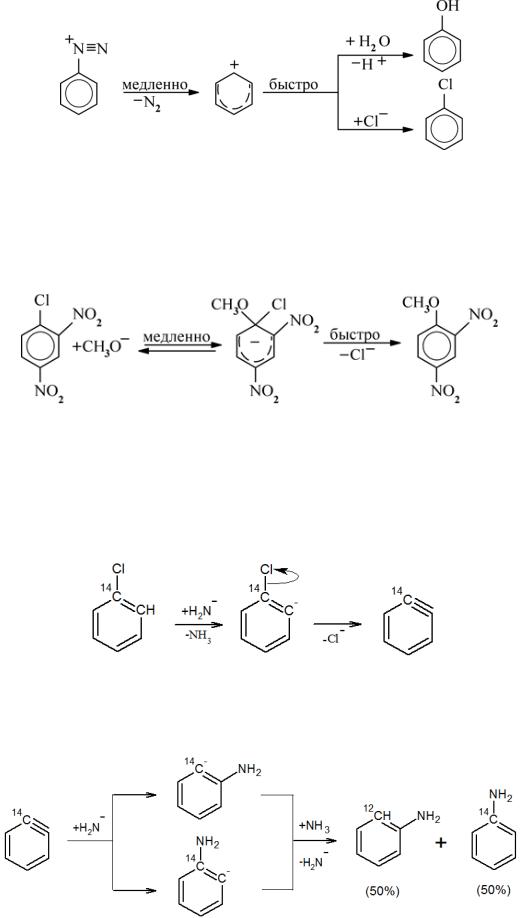
Прямое нуклеофильное замещение в ароматических соединениях, аналогичное SN2-реакции при насыщенном атоме углерода, вероятно, невозможно. Лишь при наличии сильных электроноакцепторных групп в орто‒ и пара‒положениях может протекать замещение через промежуточный анион, который стабилизируется за счет стягивания электронной плотности электроноакцепторными группами:
В большинстве случаев скорость определяющей стадией является образование аниона. При отсутствии электроноакцепторных групп замещение возможно проводить только в жестких условиях.
С очень сильными основаниями происходит первичное отщепление протона, аналогичное Е1сb-механизму:
Такой тип замещения доказали с помощью метода меченых атомов (изотоп 14С): метка распределилась поровну между углеродом при аминогруппе и углеродом, находящимся в орто-положении к ней.
47
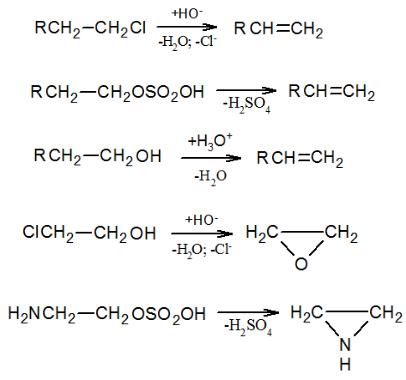
2.3. Механизм реакций отщепления
Процессы отщепления также называют реакциями элиминирования от англ. elimination. Отщепление функциональных групп может происходить из различных положений, в связи с чем различают 1,1-, 1,2-, 1,3-элиминирование и т.д. Наибольшее практическое значение имеют реакции 1,2-отщепления, в результате которых образуются соединения с двойными связями (дегидрохлорирование, дегидратация и др.), а также отдельные реакции 1,3- и 1,4-отщепления с образованием гетероциклов:
В зависимости от природы реагентов и условий проведения процесса 1,2- элиминирование может происходить по различным механизмам. Отщепление Е1 и Е2 происходят через моно- и бимолекулярные переходные состояния аналогично реакциям замещения.
При Е1-реакции вначале происходит медленный разрыв связи С–Х и образование карбкатиона. Затем под действием какого-либо нуклеофила (основание, растворитель) отрывается протон и образуется олефин:
48
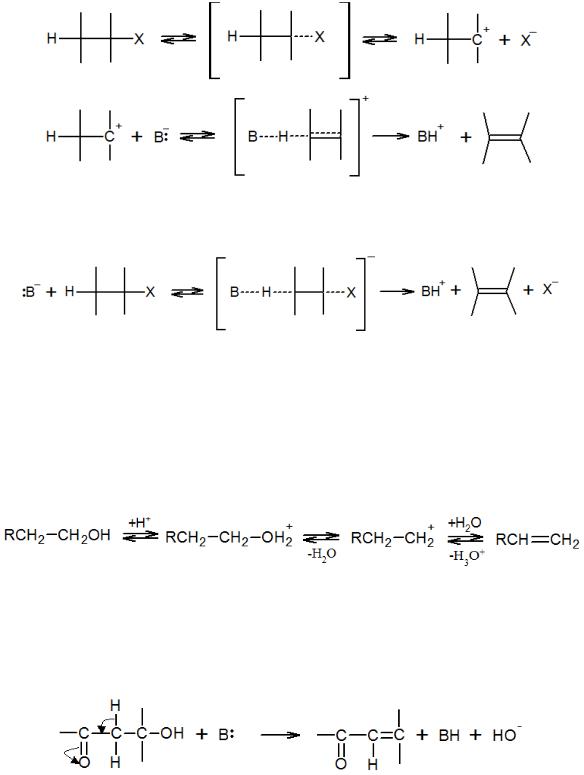
Процесс Е2-отщепления происходит путем одновременного бимолекулярного разрыва связей в молекуле, как правило, под действием сильного основания:
Факторы, влияющие на скорость отщепления в Е1- и Е2механизмах аналогичны реакциям SN1 и SN2. Мономолекулярному расщеплению способствует разветвленность алкильной группы, уменьшение энергии связи С–Х, эффекты специфической сольватации уходящей группы растворителем, наличие катализаторов. Например, дегидратация спиртов существенно ускоряется кислотами, поляризующими связь С–О:
Бимолекулярное расщепление протекает у наименее разветвленных субстратов и ускоряется электроноакцепторными заместителями. Например, дегидратация β- кетоспиртов происходит без активирования связи С–О и катализируется основаниями:
В случае нуклеофильных реакций расщепления существует третий механизм – E1cb, где индекс cb означает сопряженное основание (от англ. conjugated base). Этот путь реализуется, если чем-то сильно облегчено отщепление протона, а группа Х отщепляется трудно. Тогда вначале происходит быстрое равновесное отщепление протона, а затем медленное мономолекулярное отщепление группы Х от
49
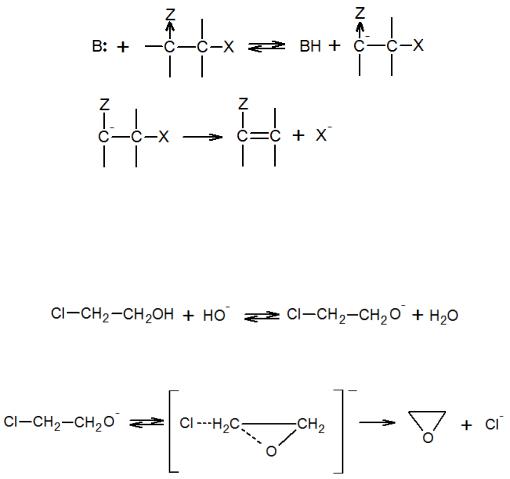
карбаниона, который является сопряженным основанием по отношению к исходному реагенту:
Электроноакцепторная группа Z увеличивает положительный заряд на уходящем атоме водорода, а также стабилизирует образующийся карбанион.
В реакции 1,3-отщеления HCl от хлоргидринов с образованием α-оксидов вначале также образуется анион
,
в котором затем происходит внутримолекулярное замещение атома хлора:
.
2.3.1. Направление отщепления
В целом, направление отщепления определяется правилом Зайцева, согласно которому реакция протекает с образованием наиболее разветвленного олефина. Определяющую роль играет строение активированного комплекса, который должен быть стабилизирован. В данном случае помогает сверхсопряжение между π- электронами образующейся двойной связи и σ-связями С-Н алкильных групп, большее количество которых снижает энергию переходного состояния и ускоряет реакцию. В соответствии с этим и будет преимущественно протекать реакция элиминирования. Например:
50