

МЕХАНИЗМЫ МЕТОДИЧКА
.pdf2.4.3. Нуклеофильное присоединение по карбонильной группе
Нуклеофильное присоединение по карбонильной группе – важный класс органических реакций, имеющих в том числе промышленное значение. К карбонильной группе способны присоединяться спирты, аммиак, амины, гидроксиламин, бисульфиты, различные кислоты и псевдокислоты (соединения, имеющие относительно подвижный атом водорода, способный замещаться на металл), к которым относятся альдегиды, кетоны, нитросоединения (а также их аналоги: нитрилы, азометины, α-метилпиридин), ацетилен.
Нуклеофильное присоединение к карбонильной группе осуществляется благодаря сильной поляризации связи С=О, которая происходит из-за смещения π- электронов в направлении более электроотрицательного атома кислорода.
C  O
O 
 C+ O-
C+ O-
Таким образом облегчается полный разрыв π-связи, причем с углеродным атомом всегда связывается нуклеофил, а с кислородным – электрофил, в частности - протон. Реакционная способность карбонильных соединений зависит от степени поляризации связи С=О. Увеличение положительного заряда на углеродном атоме способствует более легкому протеканию процесса атаки его нуклеофилом. Соответственно, с ростом числа электронодонорных заместителей реакционная способность веществ будет падать. Очевидно, что кетоны проявляют меньшую активность к нуклеофильному замещению, чем альдегиды.
Аналогичное влияние оказывают сопряженные двойные связи или функциональные группы со свободными электронными парами, повышая электронную плотность на реакционном центре ввиду сопряжения и снижая реакционную способность:
61
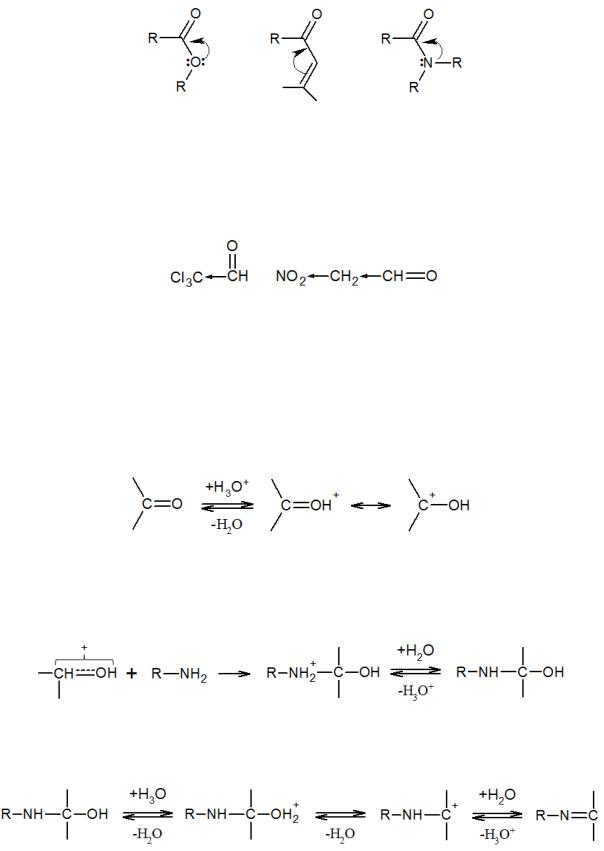
По этой причине многие α-,β-ненасыщенные кетоны, сложные эфиры, амиды и т.п. не вступают во многие реакции присоединения по карбонильной группе. Электроноакцепторные группы, напротив, ускоряют реакцию нуклеофильного присоединения, повышая положительный заряд на атоме углерода:
Поляризацию С=О связи можно усиливать с помощью добавок протонодонорных соединений, которые образуют водородные связи. Тем самым электронная плотность смещается от углерода, и он приобретает больший положительный заряд, благоприятствующий атаке нуклеофилом. В предельном случае активирования протоном полностью разрывается π-связь карбонильной группы, и образуется высокореакционноспособный карбкатион:
Это обусловливает эффективность кислотного катализа в случае присоединения основных соединений (спиртов, азотистых оснований и др.), которые присоединяются так значительно быстрее, чем некаталитически:
Кислота способствует и последующему отщеплению воды, протекающему по механизму Е1:
62
Параллельно в кислой среде протонируется и само азотистое основание с образованием катиона алкиламмония, не имеющего свободной электронной пары и, таким образом, не являющегося нуклеофилом:
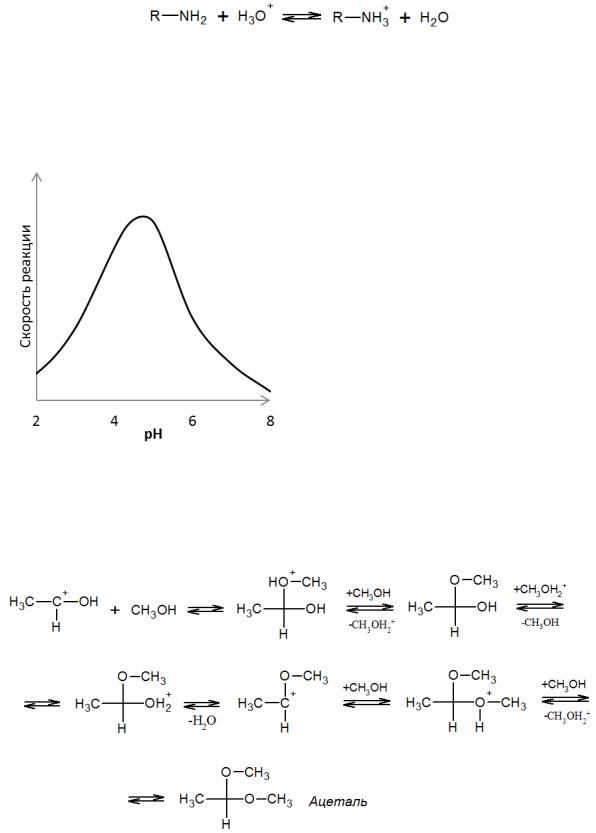
Такое противоположное влияние кислотных соединений на реакционную способность субстратов и реагентов обусловливает наличие максимума на кривой зависимости скорости нуклеофильного присоединения аминов от рН среды (рис.
2.3).
Рис. 2.3 Зависимость скорости реакции карбонильных соединений с аминами от рН среды
Аналогичным образом происходит катализируемое кислотами присоединение спиртов и меркаптанов к карбонильным соединениям, причем кислота ускоряет и дальнейшее замещение гидрокси-группы по SN1-механизму с образованием ацеталей:
63
Лимитирующей в этом процессе является стадия отщепления воды с образованием карбкатиона.
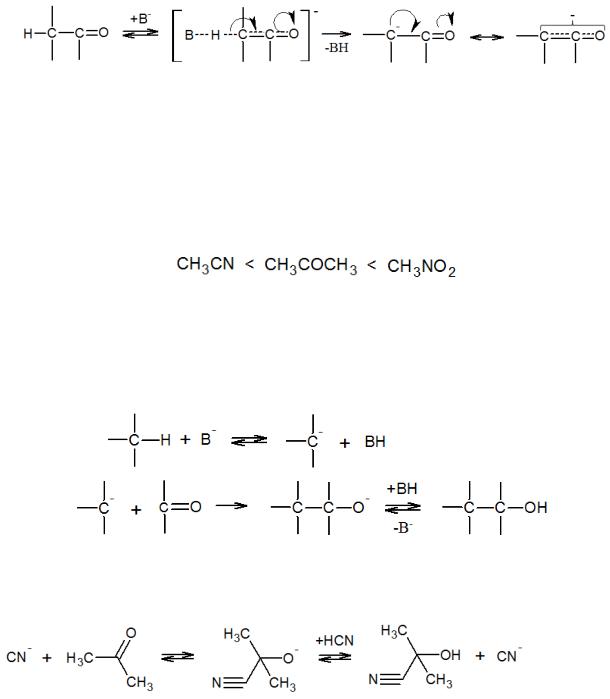
Реакции альдольной конденсации и родственные ей реакции с кислотами и псевдокислотами протекают при основном катализе. Роль катализатора заключается в образовании сопряженного реагенту основания путем бимолекулярной атаки основанием В- атома водорода при α-углеродном атоме:
Этот процесс облегчается эффектами сопряжения и делокализации электронов, стабилизирующими карбанион. Эти влияния возможны лишь при отрыве водорода от углеродного атома, находящегося в α-положении к карбонильной группе, чем и обусловлено направление реакции и строение образующихся продуктов. Поскольку катализатором является основание, скорость образования карбаниона возрастает с повышением кислотности реагента.
Образовавшийся карбанион, являющийся сильным нуклеофилом, атакует карбонильное соединение с образованием нового аниона, который отрывает протон от молекулы BH с регенерацией основного катализатора. Общая схема реакции выглядит следующим образом:
,
где роль В- и ВН выполняют HO- и H2O, NH2- и NH3 и т.д. А в реакции с синильной кислотой она сама служит донором протонов:
При конденсации двух разных карбонильных соединений в качестве карбонильного компонента всегда будет выступать наиболее реакционноспособное.
64
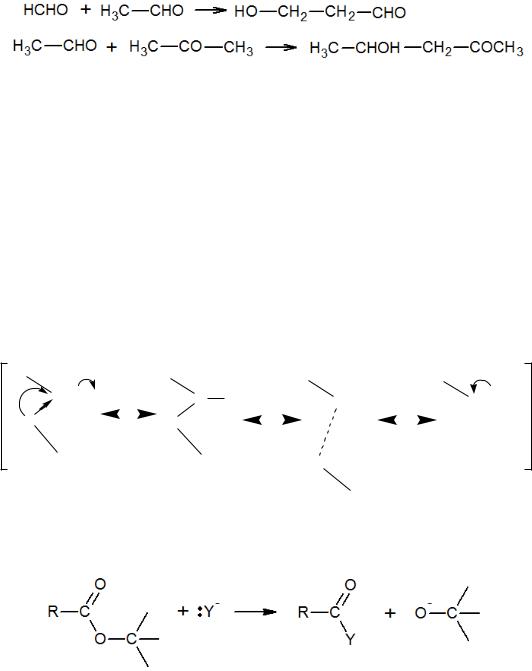
То есть формальдегид в реакциях с другими альдегидами и, тем более, с кетонами всегда будет карбонильным компонентом. А альдегиды всегда будут таковыми при взаимодействии с кетонами.
2.4.4. Нуклеофильные реакции карбоновых кислот и их производных
Ранее мы рассмотрели реакционную способность карбонильных соединений, которая в значительной степени зависит от степени поляризации связи С=О. Увеличение поляризации способствует более легкому протеканию процесса атаки атома углерода нуклеофилом. Понятно, что появление у карбонильной группы такого мощного электроноакцепторного заместителя, как ОН-группа, вызывает уже обратный эффект изменения электронной плотности по -связи, что в конечном итоге приводит к диссоциации кислоты на анион ОН- и очень реакционноспособную электрофильную ацильную группу R-C+=O.
R |
|
R + |
- |
|
R |
+ |
|
|
R |
|
|
|||
C |
|
O |
|
|
|
|
|
|
||||||
|
|
C |
O |
|
|
|
|
|
|
O |
||||
|
|
|
|
C |
|
O |
|
C |
|
|||||
|
|
|
|
|
|
|
||||||||
O |
|
O |
|
|
|
|
|
|
|
+ |
|
|||
|
|
|
|
|
|
|
||||||||
|
|
|
O - |
|
|
OH- |
||||||||
H |
|
H |
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||
H
В ходе реакций нуклеофил атакует ацильный атом углерода, и реакция протекает по следующей схеме:
Такие процессы называют ацилированием, а карбоновые кислоты и их функциональные производные – ацилирующими агентами.
Наиболее распространены реакции ацилирования, протекающие по бимолекулярному механизму. Наличие карбонильной группы обусловливает сравнительную легкость предварительного образования нестабильного продукта присоединения:
65
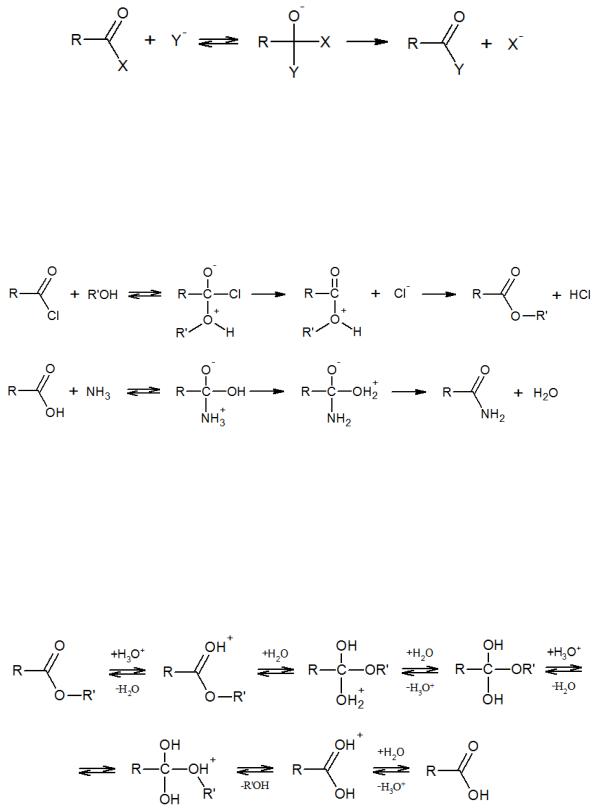
Промежуточный анион стабилизируется за счет отщепления стабильной группы Х- (RO-, NH2-, Cl-, RCOO-) с регенерацией ацильной группы. По такому механизму протекает взаимодействие кислот и сложных эфиров с аммиаком и аминами, алкоголиз сложных эфиров при катализе основаниями, нуклеофильные реакции хлорангидридов органических и неорганических кислот (фосгена, хлоридов и оксихлоридов фосфора и др.), некаталитические превращения ангидридов:
Такие реакции могут быть ускорены введением электрофильных веществ в реакционную массу, которые способны связываться с карбонильным кислородом, оттягивая на себя электронную плотность и повышая положительный заряд на атоме углерода. В этом случае применяют кислоты (в том числе кислоты Льюиса), причем наибольшая эффективность достигается за счет активирования молекулы субстрата протоном. Кислотный катализ сильно ускоряет реакции со слабыми нуклеофилами, такими, как вода или спирты:
Описанные выше реакции протекают через бимолекулярное переходное состояние, характеризующееся наличием частичного положительного заряда на реакционном центре. В связи с этим на скорость реакции будут положительно влиять эффекты заместителей, увеличивающие этот заряд. Так, последовательное
66
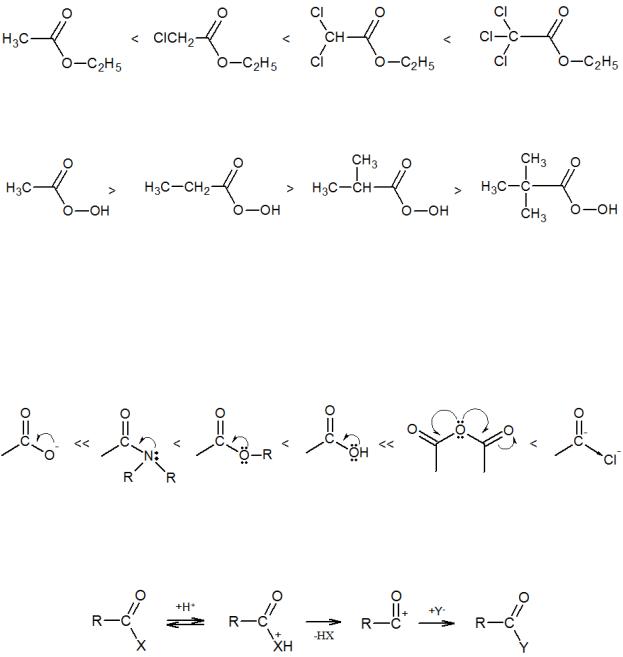
введение хлора в ацильную группу этилацетатов ускоряет их гидролиз в конечном итоге на пять порядков:
Наоборот, удлинение и разветвление углеродной цепи снижает реакционную способность карбоновых кислот и их производных:
Определенное влияние оказывает и природа замещаемой группы. Как правило, сильному отрицательному индуктивному эффекту противопоставляется положительный мезомерный эффект сопряжения свободных электронных пар гетероатома (кислород, азот), который приводит к снижению частичного положительного заряда на реакционном центре. Таким образом, имеем следующий ряд реакционных способностей:
Вприсутствии сильных кислот реакция ацилирования может идти по мономолекулярному механизму через образование ацил-катиона, который по быстрой реакции с нуклеофилом дает продукт замещения:
Вотличие от бимолекулярных превращений, в этом случае протеканию реакции способствует наличие электронодонорных заместителей, которые стабилизируют ацилкатион и облегчают его образование. По такой схеме, например, идет гидролиз метилового эфира бензойной кислоты в концентрированной серной кислоте.
Помимо вышеописанных превращений, связанных с перемещением ацильной группы из одних соединений в другие, карбоновые кислоты и их производные
67
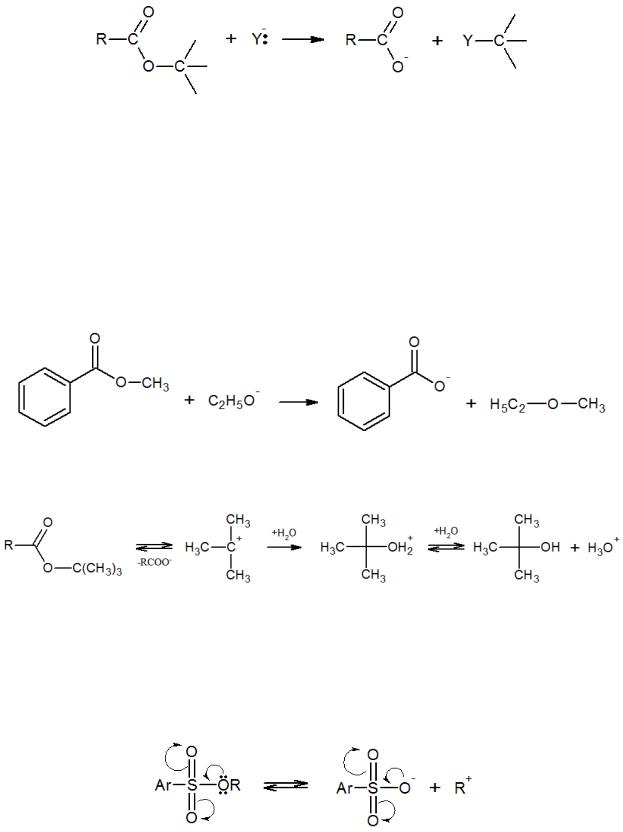
могут вступать в реакции с разрывом алкил-кислородной связи, аналогичным реакциям замещения при насыщенном атоме углерода:
Причем протекание таких реакций возможно как по моно-, так и по бимолекулярным механизмам.
Бимолекулярные реакции с разрывом связи Alk-O протекают намного медленнее, чем реакции с разрывом ацил-кислородной связи. Это связано со значительно бо́льшим дефицитом электронной плотности на ацильном углеродном атоме и с большей реакционной способностью этого положения. Примером такой реакции может служить алкоголиз метилового эфира бензойной кислоты, в результате которого образуются значительные количества простых эфиров:
По мономолекулярному механизму реагируют, например, эфиры трет- бутилового спирта:
В этом случае реакции способствуют те же факторы, что и при SN1-замещении. Одним из них является наличие электронодонорных групп, стабилизирующих промежуточный карбкатион. Другой фактор – высокая электроотрицательность кислотного остатка и возможность его стабилизации за счет сопряжения, что особенно характерно для эфиров сульфокислот и диалкилсульфатов:
68
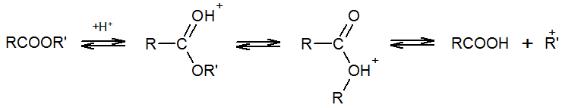
В результате такие эфиры являются сильными алкилирующими агентами. Катализируются эти реакции сильными кислотами, как и в случае
мономолекулярных реакций с разрывом ацил-кислородной связи:
Кислотный катализ способствует поляризации связи О-R’, что облегчает отщепление алкильной группы в виде карбкатиона.
3. ЭЛЕКТРОФИЛЬНЫЕ РЕАКЦИИ
Электрофильные реакции – гетеролитические реакции органических соединений с электрофильными реагентами (электрофилы, т.е. «любящие электроны»). К электрофилам относятся молекула или ее фрагмент, у которых есть свободная орбиталь на внешнем электронном уровне. Это ионы и молекулы: Н+, D+, Li+, R+, Al(R)3, Hal+, BF3, SO3H+, NO+, NO+2 и др. При взаимодействии с субстратом они принимают на себя электронную плотность. В основе многих электрофильных реакций лежит электронодонорная способность ненасыщенных соединений и ароматических углеводородов взаимодействовать с электрофилами, включая возможность передачи гетероатомами, связями С–С и С–Н своих электронных пар.
В основном органическом и нефтехимическом синтезе большое значение имеют процессы присоединения по ненасыщенными С=С связям углеводородов и реакции замещения атома или группы при атоме углерода в алифатическом ряду (substitution electrophilic - SE1 и SE2, моно- и бимолекулярные реакции, соответственно) или в ароматическом ядре - SEAr.
3.1. Электрофильное присоединение по двойным связям
Электрофильное присоединение по двойной С=С связи характерно для ряда промышленно важных реакций олефинов. Прежде всего, это реакции
69
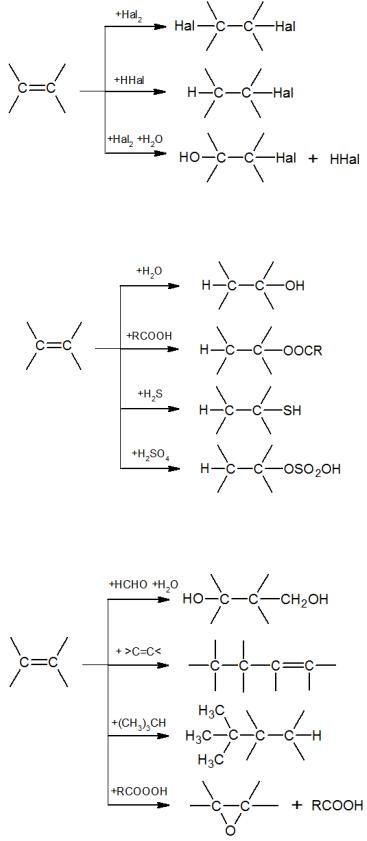
галогенирования (присоединения галогенов Hal2), гидрогалогенирования (HHal) и галогенгидринирование (хлорирование в водной среде):
Далее следуют реакции присоединения воды и кислот с получением спиртов, сложных эфиров, меркаптанов и эфиров серной кислоты:
Третья серия электрофильного присоединения к олефинам включает реакции алкилирования и эпоксидирования:
70